Recomendaciones para el reconocimiento, el diagnóstico y el manejo de este síndrome complejo y poco conocido, de consecuencias graves si no se trata.Autor/a: Dres. Danielle Cohen, Stefan P Berger, Gerda M Steup-Beekman, Kitty W M Bloemenkamp, Ingeborg M Bajeman BMJ 2010;340:c2541
El síndrome antifosfolípidos (SA) fue descrito por primera vez hace 27 años en pacientes con lupus eritematoso sistémico (LES) y presencia de anticuerpos anticardiolipina, quienes presentaban un síndrome de coagulación que afectaba las arterias y las venas. Las mujeres tenían un riesgo elevado de aborto recurrente y pérdida fetal. Los criterios internacionales de clasificación actuales se basan en las observaciones clínicas iniciales. El síndrome es poco reconocido y subdiagnosticado y si no recibe tratamiento puede tener consecuencias graves. Las dificultades diagnósticas dependen de la falta de estandarización de los análisis de laboratorio. Es muy importante la sospecha precoz, ya que el tratamiento puede reducir la mortalidad y la morbilidad en personas relativamente jóvenes que sufren enfermedades como el accidente cerebrovascular (ACV), el infarto de miocardio y la trombosis venosa profunda. Debido a esta presentación clínica variada, los pacientes con SA consultan a diferentes especialistas.
¿Qué es el síndrome antifosfolípidos?
El SA es una enfermedad sistémica autoinmune, caracterizada por trombosis arterial y venosa, complicación del embarazo (para la madre y el feto) y aumento del título de anticuerpos antifosfolípidos (AA). En más del 50% de las pacientes se presenta solo (SA primario) pero puede ir asociado a otras enfermedades inmunológicas, siendo la más común el LES—20-35% de los pacientes desarrollan un SA secundario. Una variante aguda del SA—el SA catastrófico—provoca la microangiopatía trombótica diseminada e insuficiencia multiorgánica. Los criterios de clasificación fueron actualizados por última vez en 2.006.
¿Quién los sufre?
El LES afecta de 1-20 cada 100.000 mujeres (dependiendo del origen étnico), de las que aproximadamente el 30% sufre un SA secundario. La prevalencia del SA primario en la población se desconoce, aunque se calcula que afecta al 0,5% de la población.
El SA aparece principalmente en mujeres jóvenes en edad fértil; raramente se afecta a los niños y solo en el 12% de los pacientes se presenta después de los 50 años. En un estudio de cohorte internacional, la edad media en el momento del diagnóstico fue de 34 años. La relación varón:mujer fue 1:3,5 para el SA primario y 1:7 para el SA secundario a LES. Un estudio reciente de una cohorte de 122 casos pediátricos (primarios y secundarios) informó una edad media de 10,7 al comenzar la enfermedad y una relación varón:mujer de casi 1:1,7. Los pacientes ≥50 años suelen ser varones y tener mayor prevalencia de ACV y enfermedad cardíaca coronaria. Menos del 1% de los pacientes con SA primario o secundario desarrolló una forma catastrófica y en casi la mitad de ellos apareció de novo, sin cuadros trombóticos previos.
¿Cuáles son los anticuerpos antifosfolípidos (AA) y cómo podrían causar los síntomas?
Los AA forman un grupo heterogéneo de autoanticuerpos dirigidos contra las proteínas del plasma y se unen a los fosfolípidos. Algunos anticuerpos de la familia antifosfolípidos tienen un efecto paradójico sobre la coagulación: en vivo, se asocian con trombosis recurrente pero in vitro, aumentan el tiempo de coagulación dependiente de los fosfolípidos, un fenómeno conocido como actividad “anticoagulante lúpico”. El análisis del anticoagulante lúdico es una prueba funcional basada en la combinación de varios análisis de coagulación. Para el diagnóstico de SA son útiles otros 2 anticuerpos, anticardiolipina y antiglucoproteína I β2, los cuales se detectan mediante ELISA.
Los anticuerpos con actividad anticoagulante lúpica tienen importancia clínica—dos revisiones sistemáticas recientes comprobaron su estrecha relación con las complicaciones trombóticas y obstétricas.
Los autores lamentan la falta de estandarización de esas pruebas entre los laboratorios. Una encuesta reciente que evaluó muestras plasmáticas con positividad para el anticoagulante lúdico comprobó una tasa de falsos positivos del 24%, lo que pone de relieve la importancia de una buena comunicación entre el laboratorio y la clínica y la necesidad de contar con guías.
Los AA se hallan en el 1-5% de los sujetos aparentemente sanos. Su prevalencia aumenta con la edad y puede ser influenciada por enfermedades crónicas, infecciones, cáncer y ciertos fármacos. En estas enfermedades, la positividad suele estar representada por títulos bajos de anticuerpos IgM que no se asocian con trombosis o efectos obstétricos adversos. La positividad persistente es rara. En un estudio de sección cruzada de 552 donantes de sangre sanos, el 6,5% tenía anticardiolipina IgG pero al cabo de 3 meses poco menos del 2% todavía tenía los títulos aumentados.
Para el diagnóstico definitivo de SA se requiere el cumplimiento de los criterios clínicos y la positividad de las pruebas de laboratorio en al menos 1 de 3 análisis realizados en 2 ocasiones separadas por semanas porque solo los AA persistentes tienen importancia clínica. La correlación entre los AA presentes y los síntomas clínicos es variable. Los estudios diagnósticos prospectivos con buen diseño son escasos y hay dificultad para interpretar los signos clínicos y los resultados serológicos, debido a la falta de estandarización de los ensayos, la inclusión de criterios variables y la diferencia en las definiciones existentes. En general, la evidencia avala lo siguiente:
• El anticoagulante lúpico está estrechamente relacionado con la trombosis venosa, tanto en el LES como en la población general. Este efecto se nota más en <50 años.
• El anticoagulante lúpico está estrechamente asociado con el ACV, tanto en el LES como en la población general. Este efecto es más acentuado en <5º años.
• El anticoagulante lúpico está estrechamente asociado con la pérdida fetal posterior a la 10ª semana de gestación.
• El anticoagulante lúpico predice la trombosis venosa y la pérdida fetal mejor que los anticuerpos anticardiolipina.
• Se considera que el análisis ELISA de los anticuerpos anticardiolipina tiene más sensibilidad pero menor especificidad. Un resultado positivo se correlaciona más con la morbilidad en el embarazo que con la trombosis.
• Las investigaciones sobre la relación entre los anticuerpos antiglucoproteína I ß2 y los síntomas clínicos han dado resultados contradictorios. La importancia clínica de los anticuerpos antiglucoproteína I ß2 es incierta.
• Las pacientes con positividad para los 3 anticuerpos tienen un riesgo particularmente elevado de morbilidad o tromboembolismo en el embarazo.
• Los factores de riesgo de trombosis como el tabaquismo (enfermedad arterial) y los anticonceptivos orales (trombosis venosa) aumentan más el riesgo de trombosis en presencia de AA.
• Se desconoce el riesgo en personas aparentemente sanas con AA positivos persistentes que finalmente desarrollan un cuadro clínico como la trombosis o un resultado adverso del embarazo.
Criterios de clasificación del síndrome antifosfolípidos El diagnóstico de SA se hace cuando están presentes al menos uno de los criterios clínicos y uno de los criterios de laboratorio siguientes: Criterios clínicos
Trombosis vascular Uno o más episodios de trombosis arterial, venosa o de los pequeños vasos en cualquier tejido u órgano.La trombosis debe ser confirmada por criterios objetivos válidos (imágenes o histopatología).
Confirmación histopatológico: presencia de trombosis sin signos de inflamación en la pared vascular. Morbilidad en el embarazo
Una o más muertes inexplicadas de un feto morfológicamente normal en la 10ª semana o más de gestación, con feto morfológicamente normal documentado por ecografía o examen directo.
Uno más partos prematuros de un recién nacido morfológicamente normal antes de la 34ª semana de gestación debido a eclampsia o preeclampsia grave definida por criterios estándar o un cuadro reconocido de insuficiencia placentaria.
Tres o más abortos espontáneos consecutivos inexplicados antes de la 10ª de gestación, sin anormalidades anatómicas u hormonales maternas y causas cromosómicas paternas o maternas. Criterios de laboratorio Anticoagulante lúdico en plasma en 2 o más ocasiones, con al menos 12 semanas de intervalo, detectado de acuerdo con las guías de la International Society on Thrombosis and Haemostasis.
Títulos moderado o eleados (>40 unidades de fosfolípidos IgG o Igm [1 unidad de anticuerpo es1 µg de anticuerpo]) o >percentilo 99 de anticuerpos anticardiolipina IgG o IgM en suero o plasma, en ≥2 ocasiones, con un intervalo de al menos 12 semanas , mediante ELISA. |
¿Qué se sabe acerca de su etiología y fisiopatología?
Es poco lo que se conoce sobre la causa de la producción de autoanticuerpos frente a proteínas de unión a los fosfolípidos, tales como la anti-glucoproteíona I ß 2,
Efecto sobre la coagulación y las vías inflamatorias
Los AA afectan la cascada de la coagulación y la inflamación. En un proceso mediado por la glucoproteína I ß2, los AA se unen a las plaquetas y las células endoteliales activando las células endoteliales e induciendo un estado procoagulante. La unión de los anticuerpos también activa el complemento dando como resultado el reclutamiento de otras células inflamatorias, la activación de factores tisulares, el daño endotelial y, finalmente, la trombosis. Aunque se cree que el compromiso cerebral es principalmente de naturaleza trombótica, la evidencia actual indica que los AA pueden tener efectos más directos, causando un deterioro neurológico no relacionado con la trombosis, a través de interacciones entre los anticuerpos y las células, posiblemente debido a la activación del complemento o a una solución de continuidad de la barrera hematoencefálica.
¿Existe un disparador adicional?
La mayoría de los pacientes desarrolla un cuadro trombótico aislado en algún sitio corporal, indicando que para el desarrollo de la trombosis es necesario un disparador o factor de riesgo adicional—un “segundo impacto”—siendo posibles candidatos la infección, el daño endotelial y el embarazo.
Embarazo
En un comienzo se creía que la principal causa del resultado adverso de un embarazo era la trombosis de los vasos placentarios. Sin embargo, la trombosis y el infarto de la placenta no son específicos del SA sino que ocurren en otras enfermedades como el síndrome de preeclampsia no antifosfolípidos. En los estudios in vitro y en animales se ha observado que los AA pueden unirse directamente a las células trofoblásticas y causar daño celular directo, invasividad defectuosa y una respuesta inflamatoria local, como resultado de la activación de las vías clásica y alternativa del complemento, dando cierta luz sobre la fisiopatologías de la pérdida del embarazo. Por otra parte, se ha comprobado un efecto protector de la heparina como resultado de su actividad, no solo sobre la coagulación sino también como anticomplemento. Los AA parecen causar una disfunción directa de los trofoblastos como así la activación del complemento en la interfase materno-fetal, provocando un intercambio alterado de los componentes sanguíneos entre la madre y el feto, lo cual lleva al aborto precoz, la preeclampsia, la restricción del crecimiento intrauterino o aun la muerte fetal intrauterina.
¿Cómo se presentan los pacientes con síndrome antifosfolípidos?
Las manifestaciones clínicas del SA son diversas y pueden afectar los sistemas orgánicos. La trombosis venosa, junto con las complicaciones, es más común que la trombosis arterial. En una cohorte de 1.000 pacientes, el primer síntoma fue la trombosis venosa profunda en la pierna (32%) y el embolismo (14%. Otros vasos son afectados con mayor frecuencia por la trombosis no relacionada con el SA, como las arterias renales, hepática y subclavia y, las venas retinianas, los senos cerebrales y la vena cava. Los cuadros trombóticos arteriales más comunes son el ACV y el ataque isquémico, los cuales constituyen la manifestación inicial en el 13% y 7% de los pacientes, respectivamente. Los cuadros trombóticos recurrentes con comunes. El patrón vascular de la trombosis recurrente es bastante similar en la trombosis venosa (70% de recurrencia venosa) y la trombosis arterial (90% de recurrencia arterial).
Compromiso cerebral
El compromiso cerebral es común en el SA.
La isquemia cerebral, la migraña, la disfunción cognitiva, las convulsiones, la corea, la mielitis transversa, la psicosis, la depresión y el síndrome de Guillain-Barré han sido asociados con la presencia de AA. A pesar de la estrecha relación observada entre la cefalea crónica, incluyendo la migraña, y el SA, los estudios mostraroncontradicciones. Se ha hallado una asociación entre la valvulopatía cardíaca y las manifestaciones del sistema nervioso central del síndrome, lo que indica que existe un riesgo de embolia cerebral a partir de la válvula enferma.
Compromiso de otros órganos
La anormalidad cardíaca más común en los pacientes con SA es la endocarditis bacteriana no trombótica, caracterizada por trombos de fibrina con adherencia plaquetaria sobre la superficie endotelial de las válvulas (11,6% de los pacientes durante la evolución de la enfermedad). En el 2,8% de los casos, el síntoma de presentación es el infarto de miocardio. Estudios prospectivos han mostrado que los AA se asocian con mayor riesgo de infarto de miocardio.
Condiciones que favorecen el SA Alerta roja
Trombosis venosa profunda o embolismo pulmonar inexplicados en pacientes <50 años.
ACV en <50 años.
Ataque isquémico transitorio en <50 años.
Trombosis recurrente.
Trombosis en un sitio inusual.
Pérdida fetal inexplicada luego de la 10ª semana de gestación.
Preeclampsia precoz grave.
Restricción del crecimiento intrauterino grave.
Preeclampsia con trombocitopenia grave.
Valvulopatía cardíaca (combinada con otros síntomas de este cuadro).
Diagnóstico nuevo de LES. Alerta amarilla
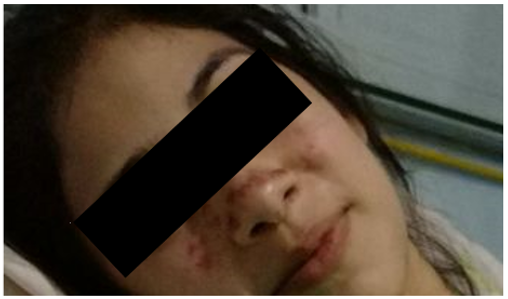
Livedo reticularis
Fenómeno de Raynaud.
Trombocitopenia persistente inexplicada.
Pérdida del embarazo precoz recurrente. |
La trombosis puede ocurrir en cualquier vaso renal. Se puede producir la oclusión de las venas renales y del tronco de la arteria renal como así microtrombos en los capilares renales, lo que puede ocasionar una disminución de la función renal. En el SA secundario no hay estudios prospectivos que hayan investigado si los AA empeoran la evolución del LES clásico, pero los análisis retrospectivos proporcionan evidencia válida para ello.
Las manifestaciones hematológicas como la trombocitopenia y la anemia hemolítica, los síntomas dérmicos, como livedo reticularis, ocurren en el 10-30% de los pacientes, aunque estos cuadros no están incluidos en los criterios de clasificación.
Situaciones en las que está indicada la determinación de anticuerpos antifosfolípidos Trombosis.
Trombosis arterial antes de los 50 años.
Trombosis venosa no provocada, antes de los 50 años.
Trombosis recurrente.
Trombosis en un sitio inusual.
Pacientes con tanto arteriales como venosos eventos trombóticos. |
Efectos maternos y fetales durante el embarazo
Los criterios obstétricos para definir el SA son: la pérdida fetal inexplicada antes de la 10ª semana de gestación; 3 o más pérdidas fetales consecutivas antes de la 10ª semana de gestación y, preeclampsia o cuadros de insuficiencia placentaria asociados con parto prematuro de bebés morfológicamente normales, antes de la 34ª semana de gestación. Otras manifestaciones que no cumplen con los criterios diagnósticos pero que son secuelas del síndrome son la trombosis materna y la restricción del crecimiento intrauterino inexplicados. La pérdida fetal está estrechamente relacionada con la presencia de AA, en particular el anticoagulante lúpico.
Estudios prospectivos han comprobado que el anticoagulante lúpico o los títulos elevados de cardiolipina IgG aumentan el riesgo de resultados adversos recurrentes. La evidencia acerca de la asociación etiológica entre los AA y el aborto es limitada. El aborto precoz es relativamente común y tiene muchas causas, de las cuales las más probables son las anormalidades cromosómicas. Es muy posible que los estudios de observación sobre la asociación entre el SA y el aborto precoz recurrente tengan muchos factores de error, sobre todo por la inclusión de abortos esporádicos más que recurrentes. Por lo tanto, las guías internacionales aconsejan hacer la pesquisa de los AA solamente en mujeres con más de 3 abortos precoces. Las mujeres con SA tienen mayor incidencia de preeclampsia precoz grave, lo que frecuentemente provoca parto prematuro iatrogénico debido a la terminación del embarazo por causas maternas o fetales. La preeclampsia con trombocitopenia grave también puede despertar la sospecha de SA y se considera un signo de alarma.
¿En quiénes se debe buscar los anticuerpos antifosfolípidos?
Lupues eritematoso sistémico
En la fase inicial del LES se recomienda el análisis de AA, el cual debe repetirse ante la emergencia de nuevos factores de riesgo de tromboembolismo. El anticoagulante lúpico y el anticuerpo anticardiolipina persistente aumentan el riesgo de cuadros tromboembólicos en pacientes con LES. Los datos sobre AA pueden ayudar cuando es difícil interpretar la aparición de nuevos síntomas e influir en las decisiones terapéuticas.
Embarazo
Un estudio prospectivo reciente de embarazadas con solo un aborto previo espontáneo antes de la 10ª semana informó que la presencia de AA aumentó significativamente el riesgo de pérdida fetal, preeclampsia y restricción del crecimiento intrauterino. Sin embargo, luego de un solo aborto, la mayoría de los embarazos posteriores no presenta complicaciones, sin ningún tratamiento. Por lo tanto, no se aconseja hacer el análisis después de un solo aborto ni en todas las mujeres que planean un embarazo.
¿Cómo se puede tratar el síndrome antifosfolípidos?
Los agentes antitrombóticos reducen el riesgo de tromboembolismo recurrente y son la base terapéutica. Las guías actuales sobre el tratamiento del SA subdividen a los pacientes en aquellos con trombosis venosa y aquellos con SA obstétrico.
Primer episodio
Para un primer episodio de trombosis venosa no provocada o de tromboembolismo asociado a AA persistentes, se recomienda la anticoagulación prolongada con antagonistas de la vitamina K como la warfarina, para reducir el riesgo de un cuadro tromnótico recurrente. Sin embargo, si existe la posibilidad de eliminar un factor de riesgo de tromboembolismo reversible—como la cirugía, la inmovilización, la estrogenoterapia o el embarazo—no está justifica la anticoagulación indefinida.
El único estudio prospectivo que se ocupó de cuadros cerebrales mostró tasas similares de tromboembolismo recurrente y de riesgo de hemorragia en pacientes tratados con warfarina o dosis bajas de aspirina. Sin embargo, el criterio inapropiado para definir los AA limita la generalización de este estudio. En pacientes con SA y ACV, se recomienda la anticoagulación prolongada con warfarina o dosis bajas de aspirina.
Dos trabajos aleatorizados y controlados que estudiaron la prevención de la trombosis venosa recurrente y los cuadros trombóticos arteriales en adultas con SA no embarazadas compararon los resultados de la anticoagulación de alta intensidad (RIN [International Normalised Ratio] de 3.1-4) con la anticoagulación de intensidad moderada (RIN 2-3). Ambos trabajos usaron warfarina oral y comprobaron que el tratamiento de alta intensidad fue mejor para la prevención de los cuadros trombóticos. Cuando se juntaron los resultados, el riesgo de hemorragia fue algo mayor en los pacientes con anticoagulación de alta intensidad. Las limitaciones de estos estudios (los pacientes con cuadros arteriales fueron minoría y muchos pacientes elegidos al azar para alcanzar un RlN >3 no cumplieron con ese objetivo) y el hecho de que los resultados se contradicen con los resultados de estudios de observación, el objetivo terapéutico todavía sigue siendo tema de debate cuál. En la actualidad, las guías internacionales y las revisiones sistemáticas recomiendan un RIN entre 2 y 3.
Prevención de las complicaciones obstétricas
Se han propuesto varias estrategias para prevenir las complicaciones trombóticas maternas y mejorar la terminación del embarazo en mujeres con SA. Existen pocos estudios bien diseñados y los estudios poblacionales son heterogéneos, de manera que el nivel de evidencia para las opciones terapéuticas es bajo.
Prevención de las complicaciones trombóticas maternas
La warfarina atraviesa la placenta y es teratogénica en el primer trimestre del embarazo, de modo que para la tromboprofilaxis prenatal se opta por las heparinas de bajo peso molecular. Los estudios de observación han demostrado que esas heparinas son al menos tan efectivas como la heparina no fraccionada, además de ser más seguras. Cuando las mujeres con SA bajo tratamiento prolongado con warfarina por haber tenido una trombosis previa deciden concebir o el embarazo ha sido confirmado, deben pasar al tratamiento con heparina. La dosis de heparina dependerá de su historia clínica siendo conveniente la consulta con el hematólogo.
Prevención del resultado adverso del embarazo
Un metaanálisis de trabajos de intervención para abortos recurrentes (precoces) concluyó que la heparina asociada a dosis bajas de aspirina reduce la pérdida del embarazo en un 64% de los casos. No se han hecho estudios controlados y aleatorizados sobre la prevención en pacientes con antecedentes de aborto tardío, muerte fetal y restricción del crecimiento intrauterino. En tales casos, se recomienda a los clínicos hacer el tratamiento con dosis bajas de aspirina y heparina (principalmente de peso molecular bajo). En pacientes con AA y antecedentes de preeclampsia grave se indican dosis bajas de aspirina (75-80 mg, una vez por día).
No se han confirmado beneficios de los glucocorticoides, los agentes citotóxicos y la inmunoglobulina intravenosa, los que pueden ser teratogénicos.
Lupus eritematoso sistémcio
Para la prevención primaria de la trombosis y la pérdida fetal en pacientes con LES y AA se pueden indicar dosis bajas de aspirina. En las pacientes con LES no embarazadas y SA asociado a trombosis se puede considerar la anticoagulación con antagonistas de la vitamina K, la cual es efectiva para la prevención secundaria de la trombosis. En las embarazadas con LES y SA se recomienda la heparina no fraccionada o la heparina de bajo peso molecular combinadas con aspirina, lo que reduce la pérdida fetal y la trombosis.
Síndrome antifosfolípidos catastrófico
El SA catastrófico es una rara condición que amenaza la vida y se caracteriza por el rápido desarrollo de múltiples microtrombos en diversos sistemas de órganos, por lo general el cerebro, los riñones, los pulmones y la piel. La trombocitopenia, la hemólisis, la esquistocitos y la activación del sistema de coagulación son hallazgos comunes de laboratorio, de modo que la púrpura trombocitopénica trombótica, el síndrome urémico hemolítico y la coagulación intravascular diseminada son diagnósticos diferenciales importantes. La mortalidad en este síndrome se acerca al 50%.
Los datos sobre el tratamiento son limitados pero los regímenes terapéuticos actuales han reducido la mortalidad, en comparación con las series de casos históricas. Los regímenes terapéuticos con buenos resultados incluyen los anticoagulantes, las dosis elevadas de corticosteroides y, el recambio plasmático con o sin Ig intravenosa. El intercambio de plasma parece ser de particular utilidad en la microangiopatía trombótica. Los trastornos precipitantes como la infección deben ser tratados inmediatamente.
Consejos para no especialistas • El reconocimiento temprano SA ayuda a prevenir la trombosis recurrente y la morbilidad materna y fetal recurrentes.
• Un diagnóstico tardío puede causar discapacidad permanente, como resultado de la formación de trombosis sin control, o incluso la muerte.
• Realizar 3 análisis de laboratorio para este síndrome.
• Trate de obtener los primeros resultados antes de empezar los anticoagulantes, porque influye en los resultados de la prueba del anticoagulante lúpico.
• Consulte con el especialista sobre los pacientes con resultados positivo.
• El embarazo conlleva un alto riesgo, y las mujeres deben ser atendidas en centros especializados.
• Los factores tradicionales de riesgo de enfermedad cardiovascular aumentan el riesgo de trombosis en este síndrome, incluso a temprana edad.
• Proporcionar apoyo a los pacientes para dejar de fumar, perder peso, y evitar los anticonceptivos orales y la terapia hormonal sustitutiva. |
Desafíos futuros para el manejo del SA
• Todavía no se cuenta con una prueba diagnóstica segura.
• El SA simula cualquier otra enfermedad, lo que genera errores diagnósticos y frustra los esfuerzos para realizar estudios de suficiente tamaño para obtener un aval inequívoco para las estrategias de diagnóstico y tratamiento. Dejar el SA sin tratamiento ocasiona secuelas graves.
• Los autores aconsejan que cualquier paciente con sospecha de SA sea visto por un equipo multidisciplinario que incluya al hematólogo, reumatólogo, neurólogo, nefrólogo y obstetra, para un mejor diagnóstico, tratamiento y educación.
Cursos de investigación y retos del futuro
• Esclarecer la relación entre la inflamación y la trombosis en el SA.
Desentrañar los efectos de los AA sobre la hemostasia, la activación endotelial y la invasividad placentaria.
• Hallar pruebas más específicas para los AA en que se correlacionen mejor con los síntomas clínicos.
• Identificar el papel terapéutico de los nuevos anticoagulantes, preferentemente orales.
• Identificar el papel de los fármacos anti-inflamatorios (rituximab, agentes anticomplemento, estatinas).
• Realizar ensayos controlados, aleatorizados y bien diseñados en embarazadas.
Resumen de puntos básicos
• Si el SA no se trata puede causar discapacidad permanente, morbilidad materna o peri natal grave o incluso la muerte.
• Los síntomas pueden aparecer en casi todos los sistemas de órganos.
• La trombosis venosa y el ACV son las manifestaciones trombóticas más comunes.
• En el embarazo, el SA se asocia con resultados adversos maternos y fetales.
• La prueba del anticoagulante lúdico es la más útil, porque su positividad tiene una estrecha relación con las manifestaciones clínicas.
• La valvulopatía cardíaca es una manifestación clínica importante y puede contribuir al riesgo de ACV. |