Rev.Colomb.Reumatol. vol.16 no.4 Bogotá Oct./Dec. 2009
Introducción
Hasta los primeros años de la década de los sesenta, la aparición de una artropatía inflamatoria en el contexto de un paciente con antecedente de psoriasis, era considerado como una coincidencia clínica en la aparición de 2 patologías distintas: artritis reumatoide y psoriasis1. Con los trabajos de Wright en 19592 y Baker en 19633, pioneros en su época, se establecen las bases para comenzar a hablar de una entidad nueva: la artritis psoriática (AP). En 1964, el Colegio Americano de Reumatología (en ese entonces llamada Asociación Americana de Reumatismo) considera a la AP como una entidad clínica distinta, incluyéndola dentro de la clasificación de enfermedades reumáticas4.
La psoriasis es una enfermedad crónica de la piel, con características inflamatorias y de hiperproliferación cutánea que afecta al 1 a 2% de los habitantes de los Estados Unidos. Su prevalencia en la población general varía entre diferentes zonas geográficas, con datos que varían entre 0 a 11.8%. Puede aparecer a cualquier edad, pero es más común su inicio entre los 15 y 30 años. En cuanto a la distribución por género, hay una distribución uniforme entre hombres y mujeres5-9.
La AP es una artropatía inflamatoria crónica de las articulaciones periféricas, columna y entesis, asociada con la presencia de psoriasis y caracterizada por unos subtipos fenotípicamente distintos y un curso clínico variable10. Hace parte del grupo de las espondiloartropatías (EAS), junto con la espondilitis anquilosante (EA), artritis reactiva, artritis relacionada con enfermedad inflamatoria intestinal (enfermedad de Crohn y colitis ulcerativa) y espondiloartropatía no diferenciada (EASND)11-12. La prevalencia exacta de AP es desconocida y su cálculo ha resultado históricamente difícil por la falta de unos criterios diagnósticos que sean ampliamente aceptados por la comunidad médica reumatológica13. Se considera su prevalencia aproximada entre 0.04 a 0.2% de la población general y entre el 6 al 39% de los pacientes con psoriasis14-19, con una tendencia al aumento en su prevalencia general en los últimos años20. La prevalencia y en general el perfil epidemiológico de la AP presentan importantes variaciones entre diferentes países y zonas del mundo, por lo que se presume la participación de factores genéticos y medio ambientales en la patogénesis y desarrollo de la enfermedad21.
Características Clínicas de la AP
En general, los hallazgos clínicos de la AP incluyen tendinitis, entesitis, dactilitis y artritis; ésta última se presenta usualmente en un patrón mono u oligoarticular en los estadios iniciales, que gradualmente va afectando a un mayor número de articulaciones pudiendo llevar a un compromiso poliarticular22, con una predilección por las articulaciones interfalángicas distales (IFD) y el esqueleto axial23. Durante mucho tiempo se consideró que el compromiso articular en la AP era de menor severidad que el observado en la artritis reumatoide (AR), pero hoy en día sabemos que cerca del 20% de los casos de AP siguen un curso clínico severo, pues después de 2 años, 47% de los pacientes tiene al menos una erosión ósea y después de 10 años más del 50% de los pacientes tiene deformidades en por lo menos 5 articulaciones, lo cual puede llevar a secuelas funcionales con alteración en la calidad de vida24-26.
A continuación nos referiremos a cada uno de los hallazgos clínicos de esta entidad.
Compromiso articular: La AP afecta tanto al esqueleto axial como al periférico. Wright y Moll describieron 5 patrones de compromiso clínico:
- Poliarticular, con artritis simétrica, semejante a la AR (15% de los pacientes)
- Oligoarticular (4 o menos articulaciones) con artritis asimétrica (70% de los pacientes)
- Compromiso predominante de AID (5% de los pacientes)
- Predominio de espondiloartritis (5% de los pacientes)
- Artritis mutilante (5% de los pacientes).
Actualmente, se acepta que estos patrones pueden ser útiles en la clasificación de la APs, sobre todo al inicio de la enfermedad; sin embargo, un paciente puede ser clasificado en más de una categoría, ya que los patrones pueden cambiar a lo largo del tiempo27, por lo que hoy en día ya no es tan práctica su utilización. También, por la llegada de nuevos criterios diagnósticos, que revisaremos más adelante en el presente artículo.
A diferencia de la AR que característicamente se presenta en forma simétrica y poliarticular, la AP compromete las articulaciones en forma asimétrica, usualmente como una mono u oligoartritis, aunque también se puede presentar como poliartritis28, usualmente en estadios mas avanzados de la enfermedad, sin embargo, la distribución del compromiso articular tiende a seguir un patrón en forma de rayo, es decir, que es más probable la afectación de todas las articulaciones de un mismo dedo, que la misma articulación en ambas manos, lo cual es típico de la AR; esto puede explicar la tendencia a la asimetría que ocurre incluso en la forma poliarticular en la ApS. Las articulaciones IFD se ven frecuentemente afectadas, lo cual puede ser un aspecto valioso a la hora de hacer el diagnóstico diferencial con AR, pero puede ser un punto de confusión con la osteoartrosis, entidad que también compromete en forma importante a este grupo de articulaciones29. La dactilitis o dedo en salchicha, es una manifestación característica de AP, presente en cerca la mitad de los pacientes; se produce por una combinación de sinovitis de las articulaciones de los dedos afectados y tendinitis30, usualmente de los tendones flexores. Los dedos con dactilitis tienen mayor riesgo de desarrollar erosiones óseas, lo cual sugiere que la presencia de dactilitis es un factor pronóstico en cuanto a la evolución de la enfermedad31. La espondiloartritis ocurre en 40% de los pacientes con AP32, presentándose en una menor proporción con relación a la artritis periférica. Se manifiesta con inflamación de las articulaciones sacroilíacas (usualmente unilateral), con dolor lumbar bajo o dolor glúteo de características inflamatorias asociado además a rigidez matutina33. La entesitis (inflamación del tendón o el ligamento en su sitio de inserción en el hueso) es un hallazgo característico de las EAS y dentro de éstas, muy especialmente en la AP, encontrándose hasta en un 38% de los casos al momento de la presentación34. Las entesis más frecuentemente afectadas son las inserciones de las fascia plantar y el tendón de Aquiles. Otros puntos que pueden verse comprometidos incluyen los tendones del cuádriceps y patelar, la cresta ilíaca y los epicóndilos35. Un último hallazgo importante de mencionar en cuanto al compromiso articular por AP es la presencia de artritis mutilante, la cual es una forma de daño articular erosivo y deformante que afecta a las articulaciones pequeñas de las manos y los pies y está acompañada por osteolisis de las falanges. Afortunadamente, este tipo de presentación tiene una muy baja frecuencia (menos del 5%)36. Finalmente, cabe anotar que los hallazgos clínicos de psoriasis usualmente preceden a las manifestaciones articulares37 hasta en 10 años17. En 75 a 85% de los casos, la artritis es posterior al desarrollo de la psoriasis38 y en alrededor de 15% de los pacientes, la artritis puede preceder a la aparición de las lesiones psoriáticas39- 40, como se evidenció en un estudio de cohorte de Hong Kong en el que 21 de 111 pacientes (18.9%) presentaron artritis antes de la psoriasis con una media de 5.4 años (+ - 4.8, rango de 1 mes a 21 años)41.
Compromiso extra articular: Hay que recordar que la AP es una enfermedad sistémica que puede afectar varios órganos y tejidos, pero que a diferencia de otras EAS, lo hace con una baja frecuencia42. Dentro de los órganos afectados se pueden encontrar: ojo (conjuntivitis, iritis, escleritis y epiescleritis), corazón, pulmones, riñones; pero como se dijo previamente, todos con un muy bajo porcentaje de presentación. Adicionalmente, se ha encontrado una mayor prevalencia de enfermedad inflamatoria intestinal en pacientes con AP43.
Características de laboratorio: No existe ningún examen de laboratorio específico para AP44. Típicamente se ha considerado la negatividad del factor reumatoide como un criterio para considerar el diagnóstico de AP, incluso hace parte de algunos criterios de clasificación para esta entidad (Moll y Wright, Bennett, Fournié y CASPAR) pero debe tenerse en cuenta que el factor reumatoide puede encontrarse positivo por otras patologías y no puede excluir por sí solo el diagnóstico de AP45, pues en caso de encontrarse en un paciente, puede tratarse de un falso positivo para AR. En el caso de los anticuerpos anti péptido cíclico citrulinado (anti CCP), los cuales, como es conocido, tienen una alta sensibilidad y especificidad para el diagnóstico de AR, se han encontrado en 5% a 10% de pacientes con AP46-48, incluso un estudio muestra un mayor porcentaje (33% de los pacientes con artritis psoriática), sin que estos pacientes llenen criterios de clasificación del ACR para AR. Estos pacientes son considerados con riesgo de desarrollar AR en forma posterior46, pero no se ha demostrado asociación entre AP y anti CCP. Los reactantes de fase aguda como velocidad de sedimentación globular (VSG) y proteína C reactiva (PCR) se encuentran elevados en los pacientes con AP pero con una menor frecuencia y en menor valor con relación a los pacientes con AR.
Características radiológicas: Los hallazgos radiológicos más comunes en AP son: la disminución del espacio articular y las erosiones óseas que afectan las articulaciones IFD y proximales49. Característicamente, estos hallazgos son asimétricos, en la misma forma que sucede con la presentación clínica de la enfermedad. Como hallazgos distintivos de la AR, en el caso de la AP no se encuentra con tanta frecuencia osteopenia yuxta articular, y en caso de presentarse, es leve. Además, las articulaciones metacarpofalángica y de la muñeca se encuentran usualmente respetadas, lo cual no sucede en la AR. Adicionalmente se pueden encontrar erosiones y resorción ósea, que en caso de comprometer a las falanges distales puede llevar a las lesiones conocidas como en "punta de lápiz"50. Se puede encontrar sacroilitis y espondilitis con osificación paravertebral, pero estos hallazgos no son específicos de la enfermedad51. Como característica radiológica adicional a tener en cuenta, la AP puede producir cambios proliferativos adyacentes a lesiones osteolíticas y erosivas en el mismo hueso (periostitis).
Criterios Diagnósticos para AP
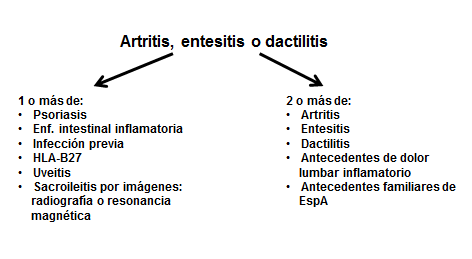
A pesar de un mayor conocimiento sobre su fisiopatología y características clínicas, la AP es una entidad que aún genera dificultades diagnósticas en la práctica clínica diaria, pudiendo llegar a constituirse en un verdadero desafío diagnóstico para el reumatólogo y aún más en el caso del médico que no tiene familiaridad con las patologías reumatológicas. Para intentar superar este problema, se han propuesto múltiples criterios de diagnóstico y clasificación a lo largo de las últimas últimas 4 décadas, (Figura 1) pero estos esfuerzos han llegado a agravar aún más la situación por la multiplicidad de criterios que pueden llegar a generar confusión entre los médicos.

Los primeros criterios propuestos fueron los de Moll y Wright52 en 1973 (sensibilidad: 91%, especificidad 98%), quienes definen a la artritis psoriática como la presencia de una artritis inflamatoria asociada a psoriasis y el factor reumatoide "generalmente" negativo (Tabla 1)



En 1991 son publicados los criterios del grupo de estudio europeo para las espondiloartropatías55, los cuales fueron propuestos originalmente para realizar una clasificación de las espondilioartritis como grupo; sin embargo, en su definición están implícitos unos criterios para la clasificación de la AP. Cabe anotar, además, que estos son los primeros criterios en los que se admite una clasificación de AP sin que haya psoriasis cutánea55 (Tabla 4).

En 1999 aparecen 2 nuevos criterios de clasificación para AP. Los propuestos por McGonagle57 y los criterios de Fournié58. Los criterios de McGonagle proponen una definición de AP basados en la entesopatía. Los criterios originales tenían a la resonancia magnética nuclear como método de evaluación radiológica de la entesitis, lo que los hacía poco prácticos con fines de tamizaje, pero el autor posteriormente propone como válida la utilización de radiografías simples para la evaluación de la entesitis (Tabla 5). Los otros criterios que aparecen al final de la década de los 90's son los de Fournié, los cuales se obtienen a partir de datos derivados de una población de pacientes diagnosticados por reumatólogos de una clínica con: AP, espondilitis anquilosante, y AR. Los componentes de estos criterios se obtuvieron a partir de datos clínicos evaluados en forma retrospectiva mediante modelos de regresión logística y análisis discriminante. Estos criterios, a diferencia de todos sus predecesores, son los únicos en utilizar una suma de puntos para establecer el diagnóstico de AP, de tal forma que con 11 puntos o más se considera este diagnóstico (Tabla 6). Cabe anotar que según la distribución de este puntaje, un paciente puede tener diagnóstico de AP sin presentar psoriasis ni artritis, así: si presenta historia familiar de psoriasis (3 puntos), factor reumatoide negativo (4 puntos) y HLA-B17 positivo (6 puntos) con lo que se alcanza la puntuación necesaria para hacer el diagnóstico.


Finalmente, en agosto del año 2006 fueron publicados los criterios de CASPAR: ClASsification criteria for Psoriatic ARthritis59 (Tabla 7). Estos nuevos criterios fueron desarrollados por un grupo internacional de investigadores con experiencia acreditada en el estudio de la AP. El estudio evaluó la utilidad de los diversos criterios existentes para la clasificación de la AP y desarrolló unos nuevos de acuerdo a los datos observados. Los datos fueron recolectados en forma prospectiva de pacientes con artritis psoriática (n=588) y controles (n=536) con otras patologías reumatológicas: AR (70%), EA(13%), artritis indiferenciada (7%), enfermedades del tejido conectivo (3%) y otras enfermedades (5%). A diferencia de los 6 anteriores criterios para la clasificación de la AP, Los criterios de CASPAR son los primeros que se basan en datos prospectivos. En cuanto a los resultados obtenidos en términos de sensibilidad y especificidad, los criterios de CASPAR no superan a algunos criterios ya existentes como los de Vasey y Espinoza, con una menor sensibilidad, aunque con mayor especificidad (Tabla 8), pero lo que contribuye a su mayor solidez es la metodología empleada en su elaboración, que los diferencia claramente de sus predecesores. En la Tabla 9 se resumen cada uno de los aspectos clínicos y paraclínicos que conforman los diferentes criterios de clasificación que hemos estudiado en el presente artículo.



Han pasado 36 años desde la aparición de los primeros criterios de clasificación de la AP y son muchos los avances que se han logrado a lo largo de este tiempo. Si bien han aparecido nuevas técnicas de imágenes diagnósticas que incluso hacen parte de algunos criterios y a pesar de la llegada de nuevas técnicas de laboratorio que han permitido tipificar el HLA, con su respectiva asociación hacia esta patología, la clínica permanece y permanecerá como pilar fundamental para llegar al diagnóstico de AP, con todas las características que el buen clínico puede encontrar a través de una adecuada anamnesis y un examen físico minucioso, detallado y adecuadamente dirigido. La multiplicidad en criterios de clasificación para AP, más que convertirse en una debilidad que pueda llevar a confusión en la práctica clínica diaria, debe llevarnos al estudio más profundo de esta entidad, aprovechando la rica experiencia de varios grupos de investigación a lo largo de los últimos años; que en últimas nos pueda conducir a una mejor aproximación diagnóstica y terapéutica hacia nuestros pacientes.
Referencias