Autor/a: Dres. Beth Goundry, Laura Bell, Matthew Langtree BMJ 2012;344:e289
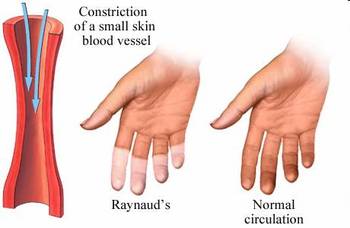
El fenómeno de Raynaud está causado por un vasoespasmo episódico y la isquemia de las extremidades en respuesta al frío o a estímulos emocionales que se traducen en un cambio de color trifásico característico, generalmente en las manos y los pies, que va del blanco, al azul y al rojo. Puede ser primario, en respuesta directa a los estímulos, o secundario a una enfermedad subyacente. En un 10-20% de los casos puede ser la primera presentación de una enfermedad del tejido conectivo o precederla en su aparición (como la esclerodermia o la enfermedad mixta del tejido conectivo), por lo que se deben descartar las causas subyacentes.
Este fenómeno es desencadenado por un cambio en la temperatura ambiente o simplemente por la exposición al frío. Los pacientes pueden tener ataques durante todo el año, por ejemplo, si se trasladan de un ambiente tibio a uno con aire acondicionado, o permanecen en el viento frío (incluso en un día relativamente cálido), o mantienen en la mano un vaso de leche fría.
Una declaración de consenso reciente de la Sección de Medicina Vascular de la Royal Society of Medicine recomienda abandonar los términos síndrome de Raynaud y enfermedad de Raynaud debido a la falta de consenso sobre su uso (Fenómeno de Raynaud: nuevos conocimientos, nuevos tratamientos. Conferencia organizada por la Sección de Medicina Vascular de la Royal Society of Medicine. Mayo de 2011).
En esta revisión los autores se refieren al fenómeno de Raynaud primario y secundario. Los avances recientes en su manejo y tratamiento surgieron de estudios aleatorizados y controlados de estrategias terapéuticas. Se revisan estudios de observación, ensayos controlados aleatorizados, revisiones sistemáticas y guías, para proporcionar una visión general de la presentación clínica, los factores de riesgo, el diagnóstico y los tratamientos.
El fenómeno de Raynaud está causado por un vasoespasmo episódico y la isquemia de las extremidades en respuesta al frío o a estímulos emocionales que se traducen en un cambio de color trifásico característico, generalmente en las manos y los pies, que va del blanco, al azul y al rojo.
¿Quiénes sufren el fenómeno de Raynaud?
La prevalencia del fenómeno de Raynaud varía ampliamente entre los países y las poblaciones. Los estudios de prevalencia no basados en la población muestran que el 3-12,5% de los hombres y el 20,6% de las mujeres relatan síntomas del fenómeno de Raynaud. La edad media de inicio es menor en las mujeres mientras que la prevalencia es más elevada en los climas fríos. En las mujeres, se asocia con la historia familiar, la exposición a los estrógenos y el estrés emocional mientras que en los hombres lo hace con el tabaquismo y el síndrome de vibración brazo-mano. El informe de la Raynaud´s and Sclerodermia Association dice que fumar más de 20 minutos reduce la temperatura corporal en 1°C.
Muchas enfermedades se han asociado con el fenómeno de Raynaud secundario, sobre todo la esclerosis sistémica y la enfermedad mixta del tejido conectivo. Un estudio de observación de casi 1.500 personas comprobó que el 89% de los casos de fenómeno de Raynaud se clasifica como primario y el 11% como secundario. Alrededor del 12,5% de los pacientes con fenómeno de Raynaud desarrolla esclerodermia y el 13,6% enfermedad del tejido conectivo.
Enfermedades asociadas con Reumatológicas
Hematológicas
Enfermedad arterial oclusiva
|
¿Cuáles son los síntomas?
Los pacientes con fenómeno de Raynaud clásico relatan cambios de color trifásicos intermitentes en las extremidades (dedos de las manos y los pies, nariz, mejillas y orejas), por lo general provocados por la exposición al frío o al estrés emocional?desde el blanco (debido a la vasoconstricción), al azul (por hipoxia tisular) y al rojo en el recalentamiento (reperfusión). Los cambios de color se asocian con sensación de opresión en las dos primeras etapas y dolor ardiente en la etapa de reperfusión.
Para hacer el diagnóstico no es necesaria la presencia de todas fases. Los cambios de color se producen de manera intermitente y tienden a desaparecer cuando los dedos se recalientan. Un ataque puede durar de minutos a horas. Los pacientes con fenómeno de Raynaud secundario probablemente padecen una enfermedad grave, la cual, si se deja sin tratamiento puede progresar a la ulceración o la gangrena.
¿Qué causa el fenómeno de Raynaud?
La fisiopatología del fenómeno de Raynaud es poco conocida y se cree que difiere entre el primario y el secundario. Se cree que en el fenómeno de Raynaud primario las anormalidades de la pared vascular son funcionales mientras que en el secundario son estructurales.
En cuanto a las anormalidades de los mecanismos de control nervioso, se considera que probablemente tienen menor importancia patogénica. Los factores intravasculares como la activación de las plaquetas y la fibrinólisis defectuosa, que reducen la capacidad de deformación de los eritrocitos y aumentan la viscosidad de la sangre se asocian con el fenómeno de Raynaud secundario mientras que la activación de los leucocitos y el estrés oxidativo se asocian tanto con el fenómeno de Raynaud primario como con el secundario. La clave para un tratamiento apropiado es comprender el mecanismo subyacente, pero todavía hay puntos que se desconocen.
¿Cómo se evalúan los pacientes con fenómeno de Raynaud?
El fenómeno de Raynaud se diagnostica clínicamente.
Historia
Interrogar a los pacientes sobre la frecuencia y el patrón de los cambios de color, en qué etapa los experimentan, qué dedos son los afectadas, las características asociadas (dolor, cambios en la sensibilidad, factores desencadenantes o que lo alivian). Para tener una imagen clara de los ataques puede ser útil llevar un diario de los síntomas. Solicitar a los pacientes que fotografíen las extremidades afectadas durante un ataque. Una investigación sistémica completa detectará las causas secundarias.
Distinción entre el fenómeno de Raynaud primario y secundario Enfermedad primaria
Enfermedad secundaria
|
Es importante preguntar al paciente si tiene alguna evidencia de erupción cutánea, fotosensibilidad, migrañas, artralgias, úlceras, disfagia y xerostomía. Las ocupaciones importantes son las que implican la exposición al frío y el uso de herramientas vibratorias. Si los empleadores no facilitan una menor exposición a dichas herramientas, los pacientes pueden tener derecho a una indemnización, según las leyes laborales locales.
Interrogar sobre los medicamentos que pueden predisponer a la aparición o el agravamiento del fenómeno de Raynaud: betabloqueantes, cloruro de vinilo, quimioterapia, derivados de la ergotamina, anfetaminas, cocaína, estrógenos (terapia de reemplazo estrogénico sin oposición, anticonceptivos orales), clonidina y simpaticomiméticos. También preguntar acerca del consumo de cigarrillos debido a que el tabaquismo lo agrava.
Para la puntuación del fenómeno de Raynaud se usa un puntaje sistemático de 0-10 que califica el nivel de dificultad experimentado y puede ayudar a determinar el impacto de la enfermedad sobre el funcionamiento del paciente.
Puntaje del fenómeno de Raynaud Interrogar al paciente sobre la frecuencia, la duración y la gravedad de los ataques para llegar a un resultado único y expresado en una escala de 0-10 (0 = paciente que no está discapacitado por los ataques; 10 = paciente extremadamente discapacitado. Preguntas
|
Examen (inspección, palpación, movilización)
 El examen debe tener en cuenta los antecedentes personales. En las manos buscar cambios de color, cambios en el lecho ungular y en la integridad de la piel. La esclerodactilia, las deformidades en flexión, las fricciones tendinosas y la calcinosis se ven en la esclerosis sistémica. Las úlceras digitales no son normales y siempre reflejan el fenómeno de Raynaud secundario; estas úlceras deben motivar enseguida el examen de otros signos de una enfermedad del tejido conectivo y la derivación al especialista.
El examen debe tener en cuenta los antecedentes personales. En las manos buscar cambios de color, cambios en el lecho ungular y en la integridad de la piel. La esclerodactilia, las deformidades en flexión, las fricciones tendinosas y la calcinosis se ven en la esclerosis sistémica. Las úlceras digitales no son normales y siempre reflejan el fenómeno de Raynaud secundario; estas úlceras deben motivar enseguida el examen de otros signos de una enfermedad del tejido conectivo y la derivación al especialista.
Palpar los pulsos periféricos. La sinovitis inflamatoria sugiere una artropatía.
Movilizar todas las articulaciones y evaluar la presencia de dolor y contractura.
Buscar la erupción malar, la alopecia no cicatrizal y las úlceras orales, los que pueden sugerir el lupus eritematoso sistémico. El descubrimiento de un endurecimiento de la piel es indicativo de esclerosis sistémica.
Identificar la sequedad de la piel, las telangiectasias y el aspecto de sal y pimienta de la hiperpigmentación y la hipopigmentación, indicativas también de la esclerosis sistémica. La presencia de livedo reticularis sugiere el lupus eritematoso sistémico o el síndrome antifosfolípidos.
Evaluar las arritmias, especialmente la fibrilación auricular y los soplos cardíacos, que están relacionados con la enfermedad tromboembólica (o en raras ocasiones, la endocarditis de Libman-Sacks). La fibrosis pulmonar hace sospechar la esclerosis sistémica.
Investigaciones
Los pacientes con fenómeno de Raynaud primario no necesitan análisis de sangre en forma sistemática. En cambio, a los pacientes con sospecha clínica de fenómeno de Raynaud secundario se les debe solicitar un hemograma completo para verificar si hay anemia y linfopenia, que están presentes en las enfermedades autoinmunes subyacentes; pruebas inmunológicas de anticuerpos antinucleares (ANA), anticuerpos antinucleares extraíbles (ENA), anti Scl-70 (topoisomerasa I), anti-Ro (SS-A), y anti-La (SS-B), marcadores inflamatorios (velocidad de eritrosedimentación, viscosidad del plasma).Los resultados negativos no excluyen una causa secundaria.
En los pacientes con signos unilaterales se debe hacer una radiografía de tórax para buscar una costilla cervical que comprima los vasos bronquiales y cefálicos. Si se sospecha un síndrome de la salida torácica está indicada la realización de una resonancia magnética.
Los exámenes especiales realizados en atención secundaria son la termografía infrarroja, la flujometría Doppler láser, la radiometría portátil y la pletismografía digital, las que muestran un patrón de cambios característico de la esclerodermia.
Además de contar con los resultados de los estudios, a menudo se hace una prueba de estimulación con frío, la cual mide la respuesta de los dedos al enfriamiento y el recalentamiento. Los dedos generalmente se recalientan en menos de 15 minutos, pero en el fenómeno de Raynaud esta fase es más larga, >20 minutos.
Si es posible, solicitar una capilaroscopia a los pacientes con sospecha de enfermedad secundaria porque con la oftalmoscopia (ampliación × 20) y la dermatoscopia (ampliación × 10) se pueden perder las alteraciones capilares. El método de oro estándar es la videocapilaroscopia (ampliación × 200, o la biomicroscopia). En los pacientes con fenómeno de Raynaud primario, la disposición de las asas capilares en el lecho de la uña es regular. En cambio, los pacientes con enfermedad de Raynaud secundaria muestran desorganización de la arquitectura, capilares gigantes, hemorragias, pérdida de capilares, angiogénesis y áreas avasculares ("patrón esclerodérmico", observado en el 95% de los casos de esclerosis sistémica). Casi el 80% de los pacientes con fenómeno de Raynaud, anticuerpos de la esclerodermia y un patrón de esclerodermia en la capilaroscopia desarrollará esclerodermia después de 15 años, pero si la capilaroscopia es normal, la probabilidad de desarrollar esclerodermia es casi nula.
En la actualidad, la capilaroscopia es parte de la nueva definición de esclerosis sistémica temprana propuesta por la European League Against Rheumatism (EULAR).
Investigaciones especializadas en atención secundaria Termografía infrarroja Flujometría láser Doppler Radiometría portable Pletismografía digital |
¿Cuándo se deben derivar los pacientes y a cuál especialista?
La mayoría de los pacientes puede ser tratada en atención primaria. Sin embargo, se puede considerar la derivación al reumatólogo si:
- El diagnóstico es dudoso.
- Se sospecha una causa secundaria.
- Se sospecha que la causa está relacionada con la ocupación laboral (derivar al servicio de medicina laboral).
- El paciente es menor de 12 años.
- Hay ulceraciones digitales.
- Hay mal control de los síntomas a pesar del tratamiento conservador apropiado.
¿Cómo se trata el fenómeno de Raynaud?
El primer paso en el manejo del fenómeno de Raynaud en atención primaria es modificar el estilo de vida. El asesoramiento respectivo se puede brindar mientras los pacientes esperan los resultados de los estudios y la derivación a la atención secundaria, independientemente de la sospecha de una causa subyacente. La mayoría de las personas con fenómeno de Raynaud primario responde bien a dichas modificaciones y no necesita tratamiento adicional. Los pacientes con fenómeno de Raynaud secundario requieren tratamiento de la enfermedad subyacente, lo que implica la derivación al especialista.
El primer paso en el manejo del fenómeno de Raynaud en atención primaria es modificar el estilo de vida.
Tratamientos no farmacológicos
Los métodos tradicionales de tratamiento están destinados a reducir la exposición a los factores desencadenantes (frío y estrés emocional). Aconsejar al paciente que trate de mantener el calor, tal vez mediante el uso de calentadores de manos y pies, disponibles en el comercio. La frecuencia y la gravedad de los ataques pueden reducirse evitando los cambios bruscos de temperatura ambiental y tomando precauciones para reducir la exposición al frío en el trabajo.
La vasodilatación puede aumentarse durante los ataques haciendo rotar los brazos como las aspas de un molino de viento, colocando las manos en agua tibia o en un pliegue del cuerpo como la axila, y mover los brazos como para nadar (levantando los dos brazos por encima de los hombros y moviéndolos con fuerza por el cuerpo para generar una fuerza que promueva el flujo de la sangre distal hacia los dedos). Otro consejo simple es evitar acarrear bolsas por las asas, lo que dificulta la circulación de los dedos. Hay pocas pruebas objetivas que indican algún beneficio a partir de suplementos nutricionales.
La reducción del estrés mediante técnicas de relajación generales puede ser beneficiosa. La biorretroalimentación ha sido un tratamiento popular, pero una revisión de Cochrane reciente halló que no es más eficaz que la biorretroalimentación simulada. Los grupos de apoyo pueden ofrecer sugerencias útiles y orientación sobre el autocuidado. Un estudio prospectivo mostró que dejar de fumar puede ayudar a reducir la gravedad pero no la ocurrencia de la enfermedad.
En los últimos 10 años se ha estudiado el ginkgo biloba. Un ensayo doble ciego controlado con placebo encontró una reducción del 56% de la frecuencia de los ataques en comparación con una reducción del 27% en el grupo placebo. Otro ensayo aleatorizado, multícéntrico, abierto, con dosis flexibles, halló una reducción del 31% en comparación con el 50,1% conseguido por la nifedipina, lo que sugiere que el ginkgo puede no ser tan eficaz como la nifedipina. Sin embargo, teniendo en cuenta que gingko no tiene efectos adversos y fue bien tolerado, puede ser de utilidad para otras investigaciones.
Tratamientos farmacológicos
Varios ensayos controlados aleatorizados en curso pueden conducir a un aumento del número de tratamientos para el fenómeno de Raynaud. Sin embargo, hasta la fecha, no se han publicado guías terapéuticas.
Los autores analizan los fármacos no aprobados para ser usados en el fenómeno de Raynaud que son utilizados actualmente para el tratamiento de esta enfermedad y que el médico puede decidir utilizar según el caso, teniendo la precaución de equilibrar la evidencia de la eficacia con la de la toxicidad. También es importante examinar los medicamentos que agravan los síntomas.
Vasodilatadores
Bloqueantes de los canales de calcio: el más utilizado para el tratamiento del fenómeno de Raynaud es la dihidropiridina, sin acción cardioselectiva. La nifedipina promueve la relajación de las células musculares lisas vasculares y por lo tanto conduce a la vasodilatación. Un meta-análisis de ensayos controlados aleatorizados comprobó que la nifedipina (10-20 mg, 3 veces/día) redujo 2,8-5,0 veces por semana el número de ataques y el 33% de la gravedad.
Sin embargo, los efectos pueden ser de corta duración, y se requieren antagonistas del calcio de acción más prolongada como la amlodipina o el diltiazem. Lamentablemente, los pacientes suelen reportar efectos adversos preocupantes como hipotensión, enrojecimiento, cefalea y taquicardia, lo que ha dado lugar a la investigación de tratamientos alternativos.
Nitratos tópicos:un estudio controlado aleatorizado de 33 pacientes comprobó que el gliceriltrinitrato tópico aplicado en el dorso del dedo provocó vasodilatación digital con menos efectos secundarios sistémicos que los nitratos por vía oral. Dos grandes ensayos clínicos recientes, aleatorizados y controlados, de MQX-503, una nueva formulación de nitroglicerina aplicada en el dedo afectado redujo la gravedad del fenómeno de Raynaud, pero no la duración o la frecuencia de los ataques. La evidencia sobre nitritos tópicos es limitada pero los resultados de los trabajos actuales pueden brindar pruebas más firmes acerca de su eficacia.
Prostaglandinas:las prostaglandinas tienen efectos vasodilatadores y antiproliferativos e inhiben la agregación plaquetaria. Sus efectos colaterales son similares a los de los bloqueantes de los canales de calcio y para cuando estos bloqueantes fracasan, la European League Against Rheumatism recomienda el uso de prostaglandinas. Los estudios con iloprost intravenoso comprobaron que reduce la frecuencia y la gravedad de los ataques. Un estudio aleatorizado mostró que el uso cíclico es beneficioso por la adherencia del paciente y su mejor calidad de vida.
Sin embargo, dos estudios aleatorizados controlados hallaron que el iloprost solo fue un poco mejor que la nifedipina. Como el iloprost es más caro, la European League Against Rheumatism aconseja a la nifedipina como medicamento de primera línea. Un estudio multicéntrico doble ciego controlado con placebo y aleatorizado halló que la administración de prostaglandinas orales a pacientes con fenómeno de Raynaud es menos efectiva que la intravenosa, aunque dosis mayores pueden brindar más beneficios. En la actualidad se está estudiando el treprostinil, un análogo de la prostaglandina oral.
Inhibidores de la fosfodiesterasa tipo 5 (sildenafil, tadalafil y vardenafil): estos inhibidores rompen el cGMP (monofosfato de guanosina cíclico) de las células endoteliales. La inhibición de esta enzima aumenta la cantidad de cGMP disponible para promover la relajación del músculo liso vascular y el flujo sanguíneo. Un estudio aleatorizado doble ciego controlado con placebo fijó la dosis durante todo el estudio, y dos series de casos hallaron una disminución de la frecuencia y gravedad de los ataques en pacientes tratados con sildenafil oral pero no con el tadalafil, en comparación con el placebo.
Estos inhibidores también mejoran el puntaje y la cicatrización de las heridas en pacientes con fenómeno de Raynaud. Los beneficios de estos fármacos administrados por vía oral y su buena tolerancia indican que puede ser un tratamiento efectivo para los pacientes con fenómeno de Raynaud grave y discapacitante, aunque se requieren más estudios para confirmarlo.
Antioxidantes: La N-acetilcisteína actúa como un vasodilatador modulando al vasodilatador adrenomedulina. Recientemente, un estudio de observación halló que los antioxidantes disminuyeron la frecuencia y la gravedad de los ataques. El número de úlceras digitales y curación de las úlceras también mejoró.
Inhibidores de la vasoconstricción
Antagonistas de los receptores de angiotensina: un estudio aleatorizado controlado mostró que el losartán reduce más la frecuencia y gravedad de los ataques que la nifedipina. La European League Against Rheumatism recomienda su uso pero esta recomendación es informal debido a que no hay evidencia suficiente.
Inhibidores de la reconversión de angiotensina: estos fármacos ya no se recomiendan debido a que un estudio aleatorizado doble ciego controlado con placebo comprobó que no reducen las úlceras digitales ni la frecuencia o gravedad de los ataques.
Bloqueantes de los adrenorreceptores α1: solo se cuenta con evidencia de bajo nivel proveniente de un estudio transversal de 24 pacientes, aleatorizado, doble ciego, controlado con placebo, que muestra que el prazosin puede reducir la frecuencia pero no la gravedad de los ataques, comparado con el placebo. Sin embargo, el prazosin raramente es utilizado para el tratamiento del fenómeno de Raynaud debido a sus potenciales efectos adversos que superan a sus beneficios.
Antagonistas de los receptores de endotelina (bosentan): la endotelina es un vasoconstrictor potente del músculo liso vascular. Entre sus acciones, ejerce un efecto constante sobre la vasculatura. El estudio Randomized Placebo Controlled Investigation of Digital Ulcers in Scleroderma (RAPIDS-1 y 2) ha mostrado que el número de úlceras digitales nuevas en pacientes con fenómeno de Raynaud secundario disminuyó significativamente cuando fueron tratadas con bosentan. La European League Against Rheumatism recomienda su uso cuando los síntomas son refractarios al tratamiento con bloqueantes de los canales de calcio y prostaglandinas.
Inhibidores de la recaptación de serotonina: se desconoce cuál es el papel exacto de estos fármacos en el tratamiento del fenómeno de Raynaud. Estos agentes bloquean la recaptación de serotonina, la cual posee acción vasoconstrictora. Un estudio piloto de 53 pacientes mostró que la fluoxetina reduce la gravedad y frecuencia de los ataques del fenómeno de Raynaud primario, comparada con la nifedipina. Su efecto en el fenómeno de Raynaud secundario es menos pronunciado. Una revisión de Cochrane de un número pequeño de estudios concluyó que otro inhibidor de la recaptación de serotonina, la ketanserina, no tuvo efectos terapéuticos beneficiosos para esta afección. Este agente puede servir en pacientes que no toleran otros fármacos debido a la hipotensión, pero se requiere mayor investigación.
Toxina botulínica A: bloquea la vasoconstricción y, aunque no hay estudios con control ciego contra placebo, los informes preliminares han sugerido que puede mejorar los síntomas, disminuir la frecuencia de los ataques y mejorar la cicatrización de las úlceras digitales.
Otros
Estatinas: luego de haber observado que las estatinas afectan la función endotelial, un estudio aleatorizado de 2008 comparó la atorvastatina con el placebo en pacientes con fenómeno de Raynaud asociado a esclerosis sistémica. Comparado con el placebo, este tratamiento redujo el número de úlceras digitales como así los marcadores endoteliales.
Aspirina: aunque no hay evidencia firme que avale su uso en pacientes con fenómeno de Raynaud, es común su administración diaria para los pacientes que no tienen contraindicaciones.
¿Es terapéutica la cirugía?
La cirugía puede estar indicada en ciertos pacientes con fenómeno de Raynaud grave y discapacidad. Las intervenciones quirúrgicas incluyen la reconstrucción arterial, la simpaticectomía periférica, la embolectomía, el desbridamiento de las úlceras o, una combinación de esas técnicas.
Hoy en día no se recomienda la simpaticectomía cervical porque los estudios de observación mostraron que no es eficaz a largo plazo y se acompaña de efectos adversos intolerables—se puede llegar a requerir la amputación digital. Un estudio terapéutico con evidencia grado III mostró que la simpaticectomía de la arteria digital (palmar) puede favorecer la cicatrización completa y disminuir el número de úlceras, aunque es un procedimiento altamente especializado. Por otra parte, este beneficio no se observa cuando la isquemia digital es crónica.
La arteriólisis descompresiva y la reconstrucción arterial pueden hacerse en forma simultánea. En la ulceración crónica y la isquemia digital crítica, el desbridamiento quirúrgico puede reducir la necesidad de amputación ante el desarrollo de una osteomielitis.
Consejos para no especialistas
|