miércoles, 22 de agosto de 2018
Amiloidosis primaria nodular cutanea asociada con psoriasis
Amiloidosis primaria nodular cuatánea asociada con psoriasis
La amiloidosis puede manifestarse como forma cutánea primaria en ausencia de compromiso de otros órganos, o como parte de una enfermedad sistémica con compromiso cutáneo secundario.
Autor: Ung CY, Carr NJ, Ardern-Jones MR. Fuente: Clin Exp Dermatol. 2014 Jul;39(5):608-11. Primary cutaneous nodular amyloidosis associated with psoriasis
La amiloidosis puede manifestarse como forma cutánea primaria en ausencia de compromiso de otros órganos, o como parte de una enfermedad sistémica con compromiso cutáneo secundario. La amiloidosis nodular cutánea primaria (PCNA) es la forma más rara de amiloidosis cutánea. Como la amiloidosis primaria sistémica, se asocia con infiltrados de inmunoglobulinas monoclonales de cadenas ligeras se piensa que se origina de infiltrados cutáneos de células plasmáticas, que típicamente rodean la lesión.
Reporte:
Se presenta una mujer de 29 años con antecedentes de un nódulo que se desarrolló sobre una cicatriz de 5 años de evolución, que aumentó lentamente de tamaño sin síntomas. Presentaba psoriasis en placas, desde los 16 años de edad, empeorando durante los últimos 7 años. Los tratamientos tópicos incluyeron corticoides, análagos de la vitamina D y formulaciones con ácido salicílico y, había requerido tratamiento con fototerapia.
Al exámen fisico se observaba un nódulo purpúrico firme, de 12x9x10 mm en la región medial derecha de la tibia (Figura 1). Los diagnósticos diferenciales incluyeron granuloma por cuerpo extraño, dermatofibroma y tumores malignos de tejidos blandos. La lesión se extirpó completamente.
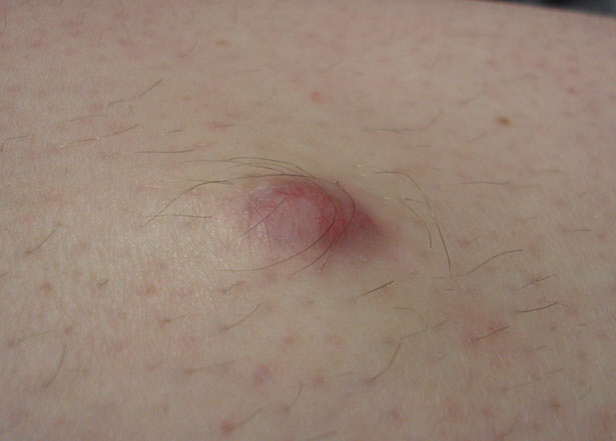
Figura 1: Nódulo en cara anterior de tibia
El exámen histológico mostró material amorfo hialino eosinofílico en dermis, reemplazando la arquitectura normal. Presentaba numerosas células gigantes, con pocas células plasmáticas (Figura 2).
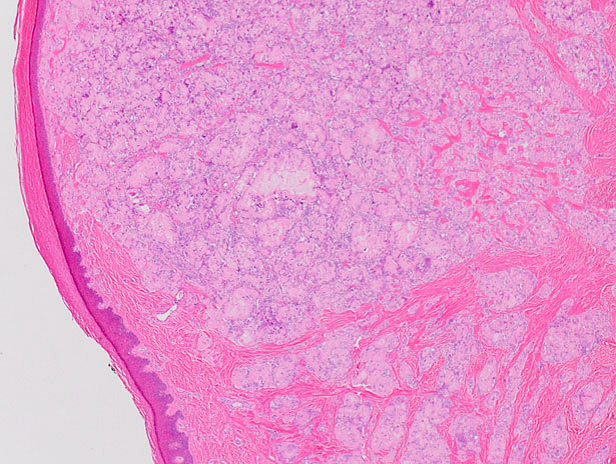
Figura 2: En dermis presentaba depósitos de amiloide, teñidos de rosa pálido
La tinción con rojo congo fue positiva, con birefringencia verde manzana al observarlo con luz polarizada de alta intensidad (Figura 3).
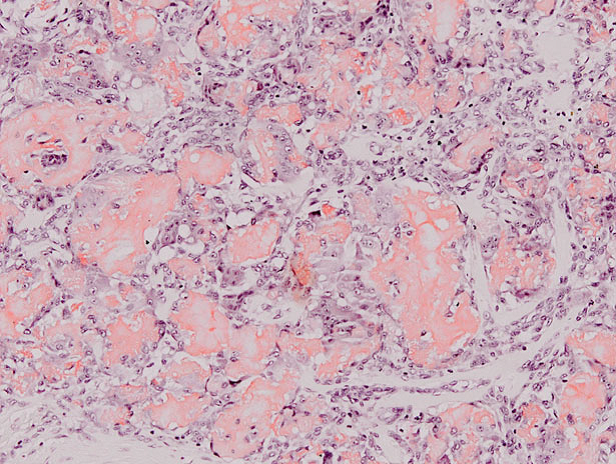
Figura 3: El depósito de amiloide fue positivo para rojo congo.
La inmunohistoquímica fue positiva para el componente amiloide P, la tinción con anticuerpos monoclonales específicos demostró reactividad a las cadenas ligeras lambda (la tinción fue negativa para cadenas livianas kappa y amiloide A).
Todas las investigaciones de laboratorio fueron normales (tabla 1). Se realizó el diagnóstico de PCNA.
Clínicamente, la PNCA se caracteriza por nódulos solitarios o múltiples, firmes, o placas infiltradas, que varían de milímetros a centímetros, con o sin epidermis atrófica suprayacente. Las áreas más comúnmente afectadas son las piernas, la cara y el tronco.
.png)
Tabla 1: Investigaciones requeridas para estudiar amiloidosis primaria cutánea y resultados en este caso.
El material hialino eosinofílico en la piel puede simular histológicamente al amiloide y, se reconoce en varias enfermedades que incluyen millium coloide, porfiria cutánea, proteinosis lipoide y macroglobulinemia de Waldenstrom.
La mayoría de las amiloidosis cutáneas primarias derivan de la apoptosis de queratinocitos, quienes típicamente liberan citoqueratina 5 (queratina amiloide; amiloide AK), pero se han identificado otras proteínas como apolipoproteína E4 (amiloide Apo E4).
Dicha amiloidosis queratínica (amiloide AK) típicamente se presenta como líquen amiloide o amiloidosis macular. La presencia de amiloide AK generalmente puede detectarse en asociación con tumores cutáneos (ej: carcinoma de células basales, carcinoma de células escamosas) y en otras situaciones (ej: queratosis seborreicas, elastosis solar, lupus discoide). Sin embargo, en este caso la proteína amiloide derivó de la producción de inmunoglobulina de cadena ligera.
La enfermedad sistémica causante de depósitos de amiloide se relaciona más comúnmente a enfermedad inflamatoria o neoplásica coexistente induciendo proteínas amiliode A (SAA amiloide), que son una familia de apolipoproteinas sintetizadas predominantemente en el hígado especialmente durante la inflamación (ej artritis reumatoidea, bronquiectasias, enfermedad de Hodgkin). Otras variantes amiloides pueden ser hereditarias y afectar proteínas específicas.
En este caso, se observó una extensa infiltración de amiloide AL en dermis, subcutis y paredes de vasos sanguíneos. Este patrón es indistinguible del encontrado en amiloidosis primaria y amiloidosis sistémica asociada a mieloma. Por lo tanto debe excluirse el compromiso sistémico en pacientes con amiloidosis nodular. Los depósitos cutáneos de amiloide AL identificados en ausencia de enfermedad sistémica son muy raros.
Sin embargo, aunque se han reportado casos de PCNA como precursores de paraproteinemia y amiloidosis sistémica, el riesgo fue de aproximadamente 7% en una serie de 15 casos. La paciente no presentó enfermedad a los 3 años de seguimiento y se realizará el screening a los 5 años nuevamente en busca de amiloidosis sistémica.
Se han descripto asociaciones de PCNA con síndrome de Sjogren, síndrome de CREST (calcicosis, fenómeno de Raynaud, enfermedades de motilidad del esófago, telangiectasia) y diabetes mellitus, pero según el conocimiento de los autores es el primer caso de asociación con psoriasis.
En este caso no se observó un infiltrado extenso de células plasmáticas, se identificó un componente histiocítico significativo. La PCNA rica y pobre en células plasmáticas ocasionan la posibilidad de que estos dos subtipos representen diferentes condiciones.
Aunque el trauma local rara vez se ha reportado como precursor, el desarrollo de PCNA, no está claro si alguno de esos pocos casos presentaba psoriasis. Es posible que el material extraño que persiste en los tejidos blandos de la injuria original podría actuar como target antigénico para la producción de anticuerpos por las células B.
Sin embargo en la inflamación de la psoriasis, los granulomas no son frecuentes, y es central la producción de interleuquina 17 por las células Th 17. Estas células han demostrado jugar un rol importante en la promoción de discrasia de células plasmáticas y mieloma, y se ha demostrado que la gammapatía monoclonal se presenta más frecuentemente en pacientes con psoriasis y artritis psoriásica.
Los autores piensan que el perfil inmunológico de la psoriasis puede haber contribuído al desarrollo de PCNA en este caso.
La proteína amiloide deriva más comúnmente de la producción aberrante de apolipoproteína y citoqueratina; la derivada de cadenas ligeras es menos común. La PCNA puede tener características similares a la amiloidosis sistémica asociada a mieloma. Los pacientes con diagnóstico de PCNA necesitan de una evaluación sistémica a largo plazo. La psoriasis se asocia con riesgo incrementado de discrasias de células plasmáticas, y este reporte sugiere una posible asociación con PCNA.
¿Qué aporta este artículo a la práctica de la dermatología?.
La amiloidosis nodular cutánea primaria (PCNA) se presenta como nódulos solitarios o múltiples firmes, con predilección por zonas acrales. Histológicamente la PCNA puede ser idéntica a la amiloidosis sistémica con depósitos de inmunoglobulina monoclonal de cadenas ligeras. La PCNA se ha asociado con otras enfermedades autoinmunes, pero según los autores esta sería la primera asociación reportada con psoriasis.
Las células T helper 17, son cruciales en la patogénesis de la psoriasis y han sido implicadas en la promoción de mieloma y discrasias de células plasmáticas. La asociación de psoriasis y producción de células plasmáticas de cadenas ligeras en la piel, sugiere el posible rol de células Th 17 en la formación de PCNA. Se discute sobre la literatura dermatopatológica de esta enfermedad rara.
Comentario y resúmen objetivo: Dra. Geraldina Rodríguez Rivello.
Reporte:
Se presenta una mujer de 29 años con antecedentes de un nódulo que se desarrolló sobre una cicatriz de 5 años de evolución, que aumentó lentamente de tamaño sin síntomas. Presentaba psoriasis en placas, desde los 16 años de edad, empeorando durante los últimos 7 años. Los tratamientos tópicos incluyeron corticoides, análagos de la vitamina D y formulaciones con ácido salicílico y, había requerido tratamiento con fototerapia.
Al exámen fisico se observaba un nódulo purpúrico firme, de 12x9x10 mm en la región medial derecha de la tibia (Figura 1). Los diagnósticos diferenciales incluyeron granuloma por cuerpo extraño, dermatofibroma y tumores malignos de tejidos blandos. La lesión se extirpó completamente.
Figura 1: Nódulo en cara anterior de tibia
Figura 2: En dermis presentaba depósitos de amiloide, teñidos de rosa pálido
Figura 3: El depósito de amiloide fue positivo para rojo congo.
Todas las investigaciones de laboratorio fueron normales (tabla 1). Se realizó el diagnóstico de PCNA.
Clínicamente, la PNCA se caracteriza por nódulos solitarios o múltiples, firmes, o placas infiltradas, que varían de milímetros a centímetros, con o sin epidermis atrófica suprayacente. Las áreas más comúnmente afectadas son las piernas, la cara y el tronco.
Tabla 1: Investigaciones requeridas para estudiar amiloidosis primaria cutánea y resultados en este caso.
La mayoría de las amiloidosis cutáneas primarias derivan de la apoptosis de queratinocitos, quienes típicamente liberan citoqueratina 5 (queratina amiloide; amiloide AK), pero se han identificado otras proteínas como apolipoproteína E4 (amiloide Apo E4).
Dicha amiloidosis queratínica (amiloide AK) típicamente se presenta como líquen amiloide o amiloidosis macular. La presencia de amiloide AK generalmente puede detectarse en asociación con tumores cutáneos (ej: carcinoma de células basales, carcinoma de células escamosas) y en otras situaciones (ej: queratosis seborreicas, elastosis solar, lupus discoide). Sin embargo, en este caso la proteína amiloide derivó de la producción de inmunoglobulina de cadena ligera.
La enfermedad sistémica causante de depósitos de amiloide se relaciona más comúnmente a enfermedad inflamatoria o neoplásica coexistente induciendo proteínas amiliode A (SAA amiloide), que son una familia de apolipoproteinas sintetizadas predominantemente en el hígado especialmente durante la inflamación (ej artritis reumatoidea, bronquiectasias, enfermedad de Hodgkin). Otras variantes amiloides pueden ser hereditarias y afectar proteínas específicas.
En este caso, se observó una extensa infiltración de amiloide AL en dermis, subcutis y paredes de vasos sanguíneos. Este patrón es indistinguible del encontrado en amiloidosis primaria y amiloidosis sistémica asociada a mieloma. Por lo tanto debe excluirse el compromiso sistémico en pacientes con amiloidosis nodular. Los depósitos cutáneos de amiloide AL identificados en ausencia de enfermedad sistémica son muy raros.
Sin embargo, aunque se han reportado casos de PCNA como precursores de paraproteinemia y amiloidosis sistémica, el riesgo fue de aproximadamente 7% en una serie de 15 casos. La paciente no presentó enfermedad a los 3 años de seguimiento y se realizará el screening a los 5 años nuevamente en busca de amiloidosis sistémica.
Se han descripto asociaciones de PCNA con síndrome de Sjogren, síndrome de CREST (calcicosis, fenómeno de Raynaud, enfermedades de motilidad del esófago, telangiectasia) y diabetes mellitus, pero según el conocimiento de los autores es el primer caso de asociación con psoriasis.
En este caso no se observó un infiltrado extenso de células plasmáticas, se identificó un componente histiocítico significativo. La PCNA rica y pobre en células plasmáticas ocasionan la posibilidad de que estos dos subtipos representen diferentes condiciones.
Aunque el trauma local rara vez se ha reportado como precursor, el desarrollo de PCNA, no está claro si alguno de esos pocos casos presentaba psoriasis. Es posible que el material extraño que persiste en los tejidos blandos de la injuria original podría actuar como target antigénico para la producción de anticuerpos por las células B.
Sin embargo en la inflamación de la psoriasis, los granulomas no son frecuentes, y es central la producción de interleuquina 17 por las células Th 17. Estas células han demostrado jugar un rol importante en la promoción de discrasia de células plasmáticas y mieloma, y se ha demostrado que la gammapatía monoclonal se presenta más frecuentemente en pacientes con psoriasis y artritis psoriásica.
Los autores piensan que el perfil inmunológico de la psoriasis puede haber contribuído al desarrollo de PCNA en este caso.
La proteína amiloide deriva más comúnmente de la producción aberrante de apolipoproteína y citoqueratina; la derivada de cadenas ligeras es menos común. La PCNA puede tener características similares a la amiloidosis sistémica asociada a mieloma. Los pacientes con diagnóstico de PCNA necesitan de una evaluación sistémica a largo plazo. La psoriasis se asocia con riesgo incrementado de discrasias de células plasmáticas, y este reporte sugiere una posible asociación con PCNA.
¿Qué aporta este artículo a la práctica de la dermatología?.
La amiloidosis nodular cutánea primaria (PCNA) se presenta como nódulos solitarios o múltiples firmes, con predilección por zonas acrales. Histológicamente la PCNA puede ser idéntica a la amiloidosis sistémica con depósitos de inmunoglobulina monoclonal de cadenas ligeras. La PCNA se ha asociado con otras enfermedades autoinmunes, pero según los autores esta sería la primera asociación reportada con psoriasis.
Las células T helper 17, son cruciales en la patogénesis de la psoriasis y han sido implicadas en la promoción de mieloma y discrasias de células plasmáticas. La asociación de psoriasis y producción de células plasmáticas de cadenas ligeras en la piel, sugiere el posible rol de células Th 17 en la formación de PCNA. Se discute sobre la literatura dermatopatológica de esta enfermedad rara.
Comentario y resúmen objetivo: Dra. Geraldina Rodríguez Rivello.
martes, 21 de agosto de 2018
Paciente con artritis y lesiones dermicas
Paciente con artritis y lesiones dérmicas

Paciente de 38 años de edad, sin antecedentes patològicos de importancia, que es derivada por su médico clínico por presentar artritis.
La paciente refirió que luego de un cuadro de anginas, desarrolla dolor de carácter inflamatorio en manos y codos. La presentación fue aditiva.
Al exámen, artritis de codo izq, con limitación a la extensión, artritis de ambos carpos, dolor a la palpación de articulaciones interfalángicas distales (DIF), artritis mediotarsial en ambos pies. No rash, no Raynaud, no caída de cabello ni úlceras mucosas.
Traía rx de manos: s/p
LABO: hto 37, bl 8500 con fórmula normal, ERS 45, PCR +++, latex-. FAN 1/80 moteado, gamma 1,99, albúmina 3,09.
SE solicitaron los anti ENA y exámen de orina y se inició tto con oh cloroquina 400 mg/dìa, deltisona 12 mg/día y Aine en caso de dolor.
En la consulta posterior: SM+, anti RNP+., hematuria de 30-40 hematíes por campo.
Al mes, la paciente desarrolla lesiones sobreelevadas pruriginosas, algo descamativas localizadas en brazos, dorso, torax y abdomen.
Se inició tto con Azatioprina 2 comp día e indico Betametasona de depósito.
La paciente refirió que luego de un cuadro de anginas, desarrolla dolor de carácter inflamatorio en manos y codos. La presentación fue aditiva.
Al exámen, artritis de codo izq, con limitación a la extensión, artritis de ambos carpos, dolor a la palpación de articulaciones interfalángicas distales (DIF), artritis mediotarsial en ambos pies. No rash, no Raynaud, no caída de cabello ni úlceras mucosas.
Traía rx de manos: s/p
LABO: hto 37, bl 8500 con fórmula normal, ERS 45, PCR +++, latex-. FAN 1/80 moteado, gamma 1,99, albúmina 3,09.
SE solicitaron los anti ENA y exámen de orina y se inició tto con oh cloroquina 400 mg/dìa, deltisona 12 mg/día y Aine en caso de dolor.
En la consulta posterior: SM+, anti RNP+., hematuria de 30-40 hematíes por campo.
Al mes, la paciente desarrolla lesiones sobreelevadas pruriginosas, algo descamativas localizadas en brazos, dorso, torax y abdomen.
Se inició tto con Azatioprina 2 comp día e indico Betametasona de depósito.
Evoluciona en forma tórpida, con leve mejoría de su artritis pero con franco empeoramiento de su cuadro dérmico.
Dicho cuadro impresionaba un LES cutáneo subagudo Vs una vasculitis urticariforme (descripta dentro de las manifestaciones del LES) Solicité toma biopsia que informó dermatitis psoriasiforme?.
40 días más tarde vuelve a la consulta presentando lesiones descamativas, pruriginosas, algo sobreelevadas localizadas en dorso, V pectoral, brazos y antebrazos. Anti DNA-, C3 y C4 normales, fan 1/1280, ers 60, PCR ++, HTO 33, BL 10800 con fórmula conservada.
El sedimento urinario mostraba escasos cilindros hialinos.
Se rota a cloroquina x2, imuran x2, prednisona 20 - 10 mg días alternos. Calcio y vitamina D.
15 días más tarde, las lesiones eran más universales. Se subió cloroquina x3 e Imuran 2 comp cada 12h.
A los 16 días ingresa al Htal Municipal de Azul, donde se interpretó el cuadro como secundario al Imuran. Se pancultivó con resultados-.
Se suspendió medicación.
Se indicó Deltisona 40 mg/día y Benadryl.
AL alta es nuevamente evaluada en consultorio. Indico nuevamente la cloroquina x2. Dejo Deltisona.
Vuelve al mes (4/01/08) con mejoría parcial de lesiones dérmicas. Inicio descenso de esteroides. Cloroquina x3. ERS 50, C3 89, C4 13. Sed urinario normal.
El día 7/3/08, la paciente se encuentra en remisión clínica. Continúa con descenso de prednisona hasta llegar a una dosis de 5 mg/día, cloroquina x2.
El anti Ro - SSA fue -, a pesar de lo que uno creería con semejante compromiso dérmico.
Recomiendo releer manifestaciones dérmicas del LES y su TTO.
Gustavo Rabazzano, Reumatólogo.
Gustavo Rabazzano, Reumatólogo.
Hombro de Milwaukee
Hombro de Milwaukee
Paciente de 76 años de edad, sexo femenino, que consulta por dolor de hombro. 
Antecedentes: HTA en tto con enalapril 10 mg/día. Sin otros antecedentes de importancia.
Consulta por dolor de carácter inflamatorio localizado en hombro derecho, de aprox. 9 meses de evolución, de comienzo insidioso, acompañado por disminución de la movilidad.
A la inspección el hombro se notaba tumefacto, se palpaba líquido en la articulación, con leve dolor a la palpación del manguito rotador. La movilidad activa y pasiva se encontraban francamente disminuídas, sobre todo la abeducción y la rotación externa.
En la rx de hombro se observaba al mismo subluxado, con erosiones en la cabeza humeral, con marcados signos artrósicos. En el laboratorio, ERS normal, FAL levemente aumentada.
Blancos normales.
Dado la edad de la paciente y los hallazgos clínicos y de imágenes, procedí a realizar artrocentesis que dió salida a líquido sinovial hemorrágico, con lo cual se arrivó a un diagnóstico clínico: Hombro de Milwaukee (hombro hemorrágico senil)
Descripción:
Esta patología es una artritis por cristales de hidroxiapatita, que se observa en ancianos, con predominio del sexo femenino. Aparentemente los cristales estimularían la producción de prostaglandina E2, estromelisina y colagenasa que inducirían la condrolisis y la lesión del aparato tendinoso que se ven en estos pacientes (como el caso que nos ocupa).
El compromiso simétrico se observa en el 60% de los pacientes. El comienzo puede ser insidioso. El dolor no siempre está presente, puede aparecer con los movimientos o ser nocturno. la movilidad está limitada, con crepitación e inestabilidad articular.
El diagnóstico se establece por la aspiración de líquido hemorrágico e identificación de cristales de hidroxiapatita mediante tinción con rojo de Alizarina. Puede hallarse cristales de pirofosfato de Ca en el 50% de los casos acompañando a los de hidroxiapatita.
En la RX se observa extenso daño en ambas superficies articulares, con artrosis secundaria.
El tto es sintomático e incluyen AINES, inyección de corticoides, plan de ejercicios, fisioterapia y aspiración articular. En casos avanzados, el tto es quirúrgico.
Dr. Gustavo Rabazzano.Especialista en Reumatología.

Antecedentes: HTA en tto con enalapril 10 mg/día. Sin otros antecedentes de importancia.
Consulta por dolor de carácter inflamatorio localizado en hombro derecho, de aprox. 9 meses de evolución, de comienzo insidioso, acompañado por disminución de la movilidad.
A la inspección el hombro se notaba tumefacto, se palpaba líquido en la articulación, con leve dolor a la palpación del manguito rotador. La movilidad activa y pasiva se encontraban francamente disminuídas, sobre todo la abeducción y la rotación externa.
En la rx de hombro se observaba al mismo subluxado, con erosiones en la cabeza humeral, con marcados signos artrósicos. En el laboratorio, ERS normal, FAL levemente aumentada.
Blancos normales.
Dado la edad de la paciente y los hallazgos clínicos y de imágenes, procedí a realizar artrocentesis que dió salida a líquido sinovial hemorrágico, con lo cual se arrivó a un diagnóstico clínico: Hombro de Milwaukee (hombro hemorrágico senil)
Descripción:
Esta patología es una artritis por cristales de hidroxiapatita, que se observa en ancianos, con predominio del sexo femenino. Aparentemente los cristales estimularían la producción de prostaglandina E2, estromelisina y colagenasa que inducirían la condrolisis y la lesión del aparato tendinoso que se ven en estos pacientes (como el caso que nos ocupa).
El compromiso simétrico se observa en el 60% de los pacientes. El comienzo puede ser insidioso. El dolor no siempre está presente, puede aparecer con los movimientos o ser nocturno. la movilidad está limitada, con crepitación e inestabilidad articular.
El diagnóstico se establece por la aspiración de líquido hemorrágico e identificación de cristales de hidroxiapatita mediante tinción con rojo de Alizarina. Puede hallarse cristales de pirofosfato de Ca en el 50% de los casos acompañando a los de hidroxiapatita.
En la RX se observa extenso daño en ambas superficies articulares, con artrosis secundaria.
El tto es sintomático e incluyen AINES, inyección de corticoides, plan de ejercicios, fisioterapia y aspiración articular. En casos avanzados, el tto es quirúrgico.
Dr. Gustavo Rabazzano.Especialista en Reumatología.
{kind=link}
lunes, 20 de agosto de 2018
Polisinovitis aguda edematosa del anciano o Sindrome RS3PE
RS3PE.

Paciente de 75 años que comienza bruscamente con un cuadro de 3 semanas de evolución que consistía en dolor e inmovilidad en cinturas escapular y pelviana, cuadro que se presentaba con más intensidad en las horas de la noche , lo que le impedía darse vuelta en la cama, y en las primeras horas de la mañana. Tenía gran dificultad en dejar la cama por la inmovilidad que el cuadro generaba, e iba mejorando con el transcurso de las horas del día a medida que aumentaba su actividad. El paciente podía precisar con exactitud el comienzo de su enfermedad y refería la hora exacta 21 días antes. Junto a este cuadro de localización proximal se agregó casi simultáneamente dolor en articulaciones metacarpofalangicas interfalangicas proximales carpos y radiocarpianas bilaterales , simétricas , con sinovitis franca al examen y con importante edema de manos sobre todo en localización dorsal con godet muy positivo(foto).
Entre sus antecedentes el paciente era un Diabético Tipo 2 insulino requirente desde hacía 3 años, que se trataba con insulina Glargina 1 dosis diaria y correcciones con insulina ultrarrápida antes de las comidas. Tenía una severa neuropatía diabética y retinopatía no proliferativa.
En el laboratorio presentaba como datos positivos leucocitosis de 12300 con 68% de segmentados y 3% en cayados, VSG de 85 mm / 1º hora, PCR 80mg/dl, FR neg Rose Ragan neg, FAN neg. El resto del laboratorio que incluía hepatograma proteinograma electroforético, calcemia monograma , perfil tiroideo era normal.
La Rx de manos, hombros, tórax pelvis fueron normales.
Se interpretó el cuadro como polimialgia reumática con compromiso articular periférico versus síndrome RS3PE (remitting seronegative symmetrical synovitis with pitting edema), se comenzó el tratamiento con Metilprednisona 20 mg y Aines con buena respuesta , que permitió bajar la dosis del esteroide rapidamente a 8 mg.
Como consecuencia de un aumento de PSA de 100 ng/ml y de una próstata aumentada de tamaño y de consistencia se realizó una biopsia prostática 11 meses después de comenzado su cuadro reumatológico que mostró Adenocarcinoma de próstata con un Gleason 3+3, con hipercaptación patológica en centelleograma óseo e nivel de pelvis, y varias vertebras columna dorsolumbar comenzando tratamiento oncológico.
Síndrome RS3PE
El síndrome RS3PE( Remitting Seronegative Symmetrical Synovitis with Pitting Edema), también llamado polisinovitis aguda edematosa del anciano fué descripto por Mc Carty en el año 1985, se caracteriza por la aparición brusca de una poliartritis simétrica seronegativa con importante edema con marcada fovea en dorso de manos. En un principio considerado un cuadro benigno cada vez son más los reportes que la asocian a síndrome paraneoplásicos sobre todo de próstata.
El tratamiento es esteroides en dosis bajas similares a los usados en Polimialgia Reumática con una remisión completa del cuadro al cabo de un año excepto en los casos paraneoplásicos.
Nuestro paciente mejoró de su cuadro reumatológico pero tuvo mala evolución desde el punto de vista oncológico, con dolores en columna y pelvis que requirieron opiáceos en dosis altas asociados a terapia anlgésica variada y tratamiento con hormonoterapia.
Finalmente el paciente presentó un cuadro brusco compatible con TEP con severo compromiso hemodinámico que le provocó el óbito
Con respecto al cuadro de RS3PE, es un cuadro de relativamente reciente descripción que debemos sospecharlo en pacientes añosos con polartritis simétricas con importante edema en manos y a veces pies y con Factor Reumatoideo negativo. Ante este cuadro estamos obligados a descartar una neoplasia oculta actual o que aparezca en la evolución
Entre sus antecedentes el paciente era un Diabético Tipo 2 insulino requirente desde hacía 3 años, que se trataba con insulina Glargina 1 dosis diaria y correcciones con insulina ultrarrápida antes de las comidas. Tenía una severa neuropatía diabética y retinopatía no proliferativa.
En el laboratorio presentaba como datos positivos leucocitosis de 12300 con 68% de segmentados y 3% en cayados, VSG de 85 mm / 1º hora, PCR 80mg/dl, FR neg Rose Ragan neg, FAN neg. El resto del laboratorio que incluía hepatograma proteinograma electroforético, calcemia monograma , perfil tiroideo era normal.
La Rx de manos, hombros, tórax pelvis fueron normales.
Se interpretó el cuadro como polimialgia reumática con compromiso articular periférico versus síndrome RS3PE (remitting seronegative symmetrical synovitis with pitting edema), se comenzó el tratamiento con Metilprednisona 20 mg y Aines con buena respuesta , que permitió bajar la dosis del esteroide rapidamente a 8 mg.
Como consecuencia de un aumento de PSA de 100 ng/ml y de una próstata aumentada de tamaño y de consistencia se realizó una biopsia prostática 11 meses después de comenzado su cuadro reumatológico que mostró Adenocarcinoma de próstata con un Gleason 3+3, con hipercaptación patológica en centelleograma óseo e nivel de pelvis, y varias vertebras columna dorsolumbar comenzando tratamiento oncológico.
Síndrome RS3PE

El síndrome RS3PE( Remitting Seronegative Symmetrical Synovitis with Pitting Edema), también llamado polisinovitis aguda edematosa del anciano fué descripto por Mc Carty en el año 1985, se caracteriza por la aparición brusca de una poliartritis simétrica seronegativa con importante edema con marcada fovea en dorso de manos. En un principio considerado un cuadro benigno cada vez son más los reportes que la asocian a síndrome paraneoplásicos sobre todo de próstata.
El tratamiento es esteroides en dosis bajas similares a los usados en Polimialgia Reumática con una remisión completa del cuadro al cabo de un año excepto en los casos paraneoplásicos.
Nuestro paciente mejoró de su cuadro reumatológico pero tuvo mala evolución desde el punto de vista oncológico, con dolores en columna y pelvis que requirieron opiáceos en dosis altas asociados a terapia anlgésica variada y tratamiento con hormonoterapia.
Finalmente el paciente presentó un cuadro brusco compatible con TEP con severo compromiso hemodinámico que le provocó el óbito
Con respecto al cuadro de RS3PE, es un cuadro de relativamente reciente descripción que debemos sospecharlo en pacientes añosos con polartritis simétricas con importante edema en manos y a veces pies y con Factor Reumatoideo negativo. Ante este cuadro estamos obligados a descartar una neoplasia oculta actual o que aparezca en la evolución
Mujer de 73años con pùrpura vasculitica,polineuropatia y compromiso renal
Mujer de 73 años con púrpura vasculítica polineuropatía y compromiso renal
Paciente de 73 años que consulta por edema y púrpura en miembros inferiores. La enfermedad había comenzado 2 meses antes cuando notó que en las últimas horas de la tarde aparecía edema bimaleolar importante. Al cabo de algunos días se agregan parestesias distales en ambos miembros inferiores, artralgias inespecíficas en pequeñas articulaciones de manos, muñecas, rodillas y lesiones elevadas de aspecto purpúrico en la misma localización por lo que consulta a su médico de cabecera.
En ese momento se objetiva una paciente con buen estado general pero que refería intensa astenia de 2 meses de evolución. Refería haber tenido hacía 4 años un cuadro diagnosticado de eritema nodoso en miembros inferiores que evolucionó bien con esteroides y Aines, pero sin demostrarse en ese momento una causa asociada. También refería ser hipertensa de moderada a severa tratada con Ramipril 5 mg, Atenolol 50 mg y Amlodipina 5 mg.
Era madre de 1 hija sana y no había antecedentes familiares de importancia. Se había desempeñado como ama de casa toda su vida.
En el examen físico se observaba edema en miembros inferiores que dejaba fóvea, lesiones purpúricas palpables de tipo petequial en ambos miembros inferiores. No se detectaron alteraciones en el examen neurológico además de ausencia del reflejo aquiliano bilateralmente a pesar de que la paciente se quejaba de parestesias y disestesias muy molestas que a veces impedían el sueño, en ambos miembros inferiores de localización distal a las rodillas.
El laboratorio mostraba según la paciente una anemia que fue estudiada por hematología se interpretó como “anemia de los trastornos crónicos” relacionada con proceso inflamatorio, infeccioso o vasculítico, leucocitos 9800 con fórmula normal, TGO 190 TGP 240, VSG 55 mm, creatinina 0,8mg% , urea 35 mg%, FAN+ 1/1280 patrón moteado, Anti DNA neg, Latex AR neg, Anti RO (SS-A) y Anti LA(SS-B) negativos, Anticentrómero neg, Anti Jo neg, ANCA p positivo 1/160,C3 1353(90-180) C4 23(10-40) CH50 32(25-50) Anti DNA 172(<200), TSH 7,1. Sedimento de orina: campo cubierto de eritrocitos dismórficos, cilindros hialinos y glóbulos de grasa.
Se medicó con Levotiroxina 50 ug y Metilprednisona 60 mg descendiendo rápidamente la dosis a 10 mg, con lo cual obtuvo alivio sintomático y desaparición de las lesiones purpúricas.
Al cabo de 3 meses la paciente suspende los corticoides por su cuenta notando nuevamente la aparición de edema importante en miembros inferiores, así como púrpura de tipo petequial en miembros inferiores por lo que decide consultar.
En la consulta se observaba una paciente en buen estado general que refería astenia importante, artralgias generalizadas y edema en miembros inferiores con, exantema purpúrico de tipo petequial en ambos miembros inferiores. Estas últimas lesiones tenían, algunas de ellas un borde sobreelevado(púrpura palpable).
Se realizaron nuevos análisis de laboratorio que siguieron mostrando alteraciones en el hepatograma por lo que se le solicitó investigación de virus hepatotropos. Hbs Ag neg, HBc IgG NEG, investigac de HCV positivo, con RNA viral positivo por PCR en títulos altos > 11 (índice de positividad <1). Investigación de virus de Epstein Barr y CMV negativos.
Un nuevo test para FR fue positivo por lo que se solicitó la investigación de Crioglobulinas siendo estas últimas positivas.
Se interpretó el cuadro com Crioglobulinemia Mixta Esencial secundaria a infección HCV
CRIOGLOBULINEMIA
Las Crioglobulinas son inmunoglobulinas que precipitan en forma reversible a bajas temperaturas
Clásicamente se clasificaron en tres tipos:
1) Tipo I o Crioglobulinemia simple generalmente una IgM monoclonal aunque puede ser menos frecuentemente una IgG IgA o cadenas livianas.
2) TipoII y III o Crioglobulinemia Mixta que contienen generalmente Factor Reumatoideo(usualmente una IgM) y raramente IgG o IgA. Esos Factores Reumatoideos forman complejos con la porción cristalizable(Fc) de una IgG
Las Crioglobulinemias también pueden ser clasificadas basándose en la asociación del síndrome con la enfermedad de base. Cuando no hay asociación con ninguna enfermedad de base ha sido conocida como CrioglobulinemiaEsencial o Idiomática. Sin embargo el descubrimiento de la cercana asociación entre Virus de Hepatitis C y Crioglobulinemia ha puesto dudas sobre el término de Crioglobulinemia Mixta Esencial, ya que todo este grupo de entidades serían en la actualidad consecuencia de la infección por el HCV. Las Crioglobulinemias asociadas con enfermedades particulares(trastornos Linfoproliferativos, enfermedades autoinmunes, enfermedades infecciosas), son conocidas como Crioglobulinemias Secundarias.
El mecanismo de crioprecipitación es mal comprendido pero la precipitación de estas proteinas produciría daño vasculítico. Algunas secuelas de la Crioglobulinemia es considerada relacionada a enfermedad por inmunocmplejos(vg Glomerulonefritis, Vasculitis crónica). Otras secuelas están relacionadas con la crioprecipitación, “taponaje” o trombosis, de pequeñas arterias y capilares en las extremidades(gangrena), o en los glomerulos(fallo renal agudo). Grandes crioproteinas circulantes pueden conducir al Síndrome de Hiperviscosidad)
La Crioglobulinemia Tipo I puede resultar en Hiperviscosidad debido a altos niveles de crioglobulinasmonoclonales circulantes conduciendo a obstrucción de los vasos. La concentración puede alcanzar 8g/L. Además puede haber daño no obstructivo, ocasionado por deposición de inmunocomplejos y vasculitis subsecuente.
La Crioglobulinemias Tipo II y III están asociadas con estados inflamatorios crónicos como el LES, Sjogren e infección viral por HCV. En estos trastornosLa fracción IgGes siempre policlonal con con IgM monoclonal (TipoII) o policlonal (Tipo III)
Las manifestaciones clínicas asociadas con la Crioglobulinemia TipoI , están relacionadas con la Hiperviscosidad y la Trombosis e incluyen Raynaud con ulceración digital, retinal, hemorragias severas, acrocianosis, livedo reticularis y trombosis arterial.
Las manifestaciones asociadas a la Crioglobulinemia Tipo II y III incluyen compromiso articular(usualmente artralgias de interfalanficas proximales, metacarpofalangicas, rodillas tobillos) fatiga, mialgias, enfermedad renal por complejos inmunes, vasculitis cutaneas y neuropatía periférica. Todo lo que tenía nuestra paciente. El compromiso renal es la mas seria manifestación y casi siempre obedece a una Glomerulonefritis Membranoproliferativa.
La Clásica tríada de Meltzer (purpura, artralgia, y debilidad) fue descripta por Mltzer y Franklin en 1966 se ve en el 25 a 30% de los pacientes con Crioglobulinemia Tipo II o III
En el Laboratorio la muestra de sangre debe ser tomada a 37ºC sin anticoagulantes, permitir que la sangre coagule antes de remover el suero con centrifugación a 37ºC. Luego se coloca el suero entubo a 4ºC y se incuba un tiempo variable, que en el caso de la Crioglobulinemia TipoI será de 24 hs o has ta 7 dias en el caso de las Crioglobulinemia tipo III).
La eritrosedimentación elevada puede ser por la formación de rouleaux. Puede haber hipocomplementemia
La presencia de FAN y otras manifestaciones de autoinmunidad es un fenómeno muy conocido en la Hepatitis C asi como la presncia de ANCA p.
El tratamiento es el de la enfermedad de base , asi com evitar la precipitación de las Crioglobulinas para evitar su efecto obstructivo e inflamatorio.
Los AINES se pueden usar en pacientes con artralgias y fatiga, y los inmunosupresores(corticoides, ciclofosfamida o azatioprina) están indicados cuando hay evidencia de compromiso orgánico tal como vasculitis, enfermedad renal progresiva, trastornos neurológicos o manifestaciones dérmicas severas. La plasmaféresis se indica en complicaciones que ponen en peligro la vida como crioprecipitación o hiperviscosidad. El PEG Interferon alfa combinado con Rivabirina ha demostrado eficacia en pacientes con Crioglobulinemia asociada a Hepatitis C. Pequeños ensayos con Rituximab (Ac monoclonal quimérico anti CD20), ha mostrado efectividad
Nuestra paciente tuvo una mejoría espontánea en el seguimiento. No se llevó a cabo biopsia hepática por tratarse de una paciente de mas de 70 años. La actividad inflamatoria hepática ha permanecido silente desde el punto de vista de laboratorio, y mejoraron concomitantemente las lesiones de piel asi com el compromiso renal. Nunca se vió afectada la función renal y el clearence de creatinina fue siempre normal para la edad. El sedimento de orina se normalizó y la única manifestación que ha persistido en el tiempo luego de 4 años de seguimiento es la neuropatía periférica
En ese momento se objetiva una paciente con buen estado general pero que refería intensa astenia de 2 meses de evolución. Refería haber tenido hacía 4 años un cuadro diagnosticado de eritema nodoso en miembros inferiores que evolucionó bien con esteroides y Aines, pero sin demostrarse en ese momento una causa asociada. También refería ser hipertensa de moderada a severa tratada con Ramipril 5 mg, Atenolol 50 mg y Amlodipina 5 mg.
Era madre de 1 hija sana y no había antecedentes familiares de importancia. Se había desempeñado como ama de casa toda su vida.
En el examen físico se observaba edema en miembros inferiores que dejaba fóvea, lesiones purpúricas palpables de tipo petequial en ambos miembros inferiores. No se detectaron alteraciones en el examen neurológico además de ausencia del reflejo aquiliano bilateralmente a pesar de que la paciente se quejaba de parestesias y disestesias muy molestas que a veces impedían el sueño, en ambos miembros inferiores de localización distal a las rodillas.
El laboratorio mostraba según la paciente una anemia que fue estudiada por hematología se interpretó como “anemia de los trastornos crónicos” relacionada con proceso inflamatorio, infeccioso o vasculítico, leucocitos 9800 con fórmula normal, TGO 190 TGP 240, VSG 55 mm, creatinina 0,8mg% , urea 35 mg%, FAN+ 1/1280 patrón moteado, Anti DNA neg, Latex AR neg, Anti RO (SS-A) y Anti LA(SS-B) negativos, Anticentrómero neg, Anti Jo neg, ANCA p positivo 1/160,C3 1353(90-180) C4 23(10-40) CH50 32(25-50) Anti DNA 172(<200), TSH 7,1. Sedimento de orina: campo cubierto de eritrocitos dismórficos, cilindros hialinos y glóbulos de grasa.
Se medicó con Levotiroxina 50 ug y Metilprednisona 60 mg descendiendo rápidamente la dosis a 10 mg, con lo cual obtuvo alivio sintomático y desaparición de las lesiones purpúricas.
Al cabo de 3 meses la paciente suspende los corticoides por su cuenta notando nuevamente la aparición de edema importante en miembros inferiores, así como púrpura de tipo petequial en miembros inferiores por lo que decide consultar.
En la consulta se observaba una paciente en buen estado general que refería astenia importante, artralgias generalizadas y edema en miembros inferiores con, exantema purpúrico de tipo petequial en ambos miembros inferiores. Estas últimas lesiones tenían, algunas de ellas un borde sobreelevado(púrpura palpable).
Se realizaron nuevos análisis de laboratorio que siguieron mostrando alteraciones en el hepatograma por lo que se le solicitó investigación de virus hepatotropos. Hbs Ag neg, HBc IgG NEG, investigac de HCV positivo, con RNA viral positivo por PCR en títulos altos > 11 (índice de positividad <1). Investigación de virus de Epstein Barr y CMV negativos.
Un nuevo test para FR fue positivo por lo que se solicitó la investigación de Crioglobulinas siendo estas últimas positivas.
Se interpretó el cuadro com Crioglobulinemia Mixta Esencial secundaria a infección HCV
CRIOGLOBULINEMIA
Las Crioglobulinas son inmunoglobulinas que precipitan en forma reversible a bajas temperaturas
Clásicamente se clasificaron en tres tipos:
1) Tipo I o Crioglobulinemia simple generalmente una IgM monoclonal aunque puede ser menos frecuentemente una IgG IgA o cadenas livianas.
2) TipoII y III o Crioglobulinemia Mixta que contienen generalmente Factor Reumatoideo(usualmente una IgM) y raramente IgG o IgA. Esos Factores Reumatoideos forman complejos con la porción cristalizable(Fc) de una IgG
Las Crioglobulinemias también pueden ser clasificadas basándose en la asociación del síndrome con la enfermedad de base. Cuando no hay asociación con ninguna enfermedad de base ha sido conocida como CrioglobulinemiaEsencial o Idiomática. Sin embargo el descubrimiento de la cercana asociación entre Virus de Hepatitis C y Crioglobulinemia ha puesto dudas sobre el término de Crioglobulinemia Mixta Esencial, ya que todo este grupo de entidades serían en la actualidad consecuencia de la infección por el HCV. Las Crioglobulinemias asociadas con enfermedades particulares(trastornos Linfoproliferativos, enfermedades autoinmunes, enfermedades infecciosas), son conocidas como Crioglobulinemias Secundarias.
El mecanismo de crioprecipitación es mal comprendido pero la precipitación de estas proteinas produciría daño vasculítico. Algunas secuelas de la Crioglobulinemia es considerada relacionada a enfermedad por inmunocmplejos(vg Glomerulonefritis, Vasculitis crónica). Otras secuelas están relacionadas con la crioprecipitación, “taponaje” o trombosis, de pequeñas arterias y capilares en las extremidades(gangrena), o en los glomerulos(fallo renal agudo). Grandes crioproteinas circulantes pueden conducir al Síndrome de Hiperviscosidad)
La Crioglobulinemia Tipo I puede resultar en Hiperviscosidad debido a altos niveles de crioglobulinasmonoclonales circulantes conduciendo a obstrucción de los vasos. La concentración puede alcanzar 8g/L. Además puede haber daño no obstructivo, ocasionado por deposición de inmunocomplejos y vasculitis subsecuente.
La Crioglobulinemias Tipo II y III están asociadas con estados inflamatorios crónicos como el LES, Sjogren e infección viral por HCV. En estos trastornosLa fracción IgGes siempre policlonal con con IgM monoclonal (TipoII) o policlonal (Tipo III)
Las manifestaciones clínicas asociadas con la Crioglobulinemia TipoI , están relacionadas con la Hiperviscosidad y la Trombosis e incluyen Raynaud con ulceración digital, retinal, hemorragias severas, acrocianosis, livedo reticularis y trombosis arterial.
Las manifestaciones asociadas a la Crioglobulinemia Tipo II y III incluyen compromiso articular(usualmente artralgias de interfalanficas proximales, metacarpofalangicas, rodillas tobillos) fatiga, mialgias, enfermedad renal por complejos inmunes, vasculitis cutaneas y neuropatía periférica. Todo lo que tenía nuestra paciente. El compromiso renal es la mas seria manifestación y casi siempre obedece a una Glomerulonefritis Membranoproliferativa.
La Clásica tríada de Meltzer (purpura, artralgia, y debilidad) fue descripta por Mltzer y Franklin en 1966 se ve en el 25 a 30% de los pacientes con Crioglobulinemia Tipo II o III
En el Laboratorio la muestra de sangre debe ser tomada a 37ºC sin anticoagulantes, permitir que la sangre coagule antes de remover el suero con centrifugación a 37ºC. Luego se coloca el suero entubo a 4ºC y se incuba un tiempo variable, que en el caso de la Crioglobulinemia TipoI será de 24 hs o has ta 7 dias en el caso de las Crioglobulinemia tipo III).
La eritrosedimentación elevada puede ser por la formación de rouleaux. Puede haber hipocomplementemia
La presencia de FAN y otras manifestaciones de autoinmunidad es un fenómeno muy conocido en la Hepatitis C asi como la presncia de ANCA p.
El tratamiento es el de la enfermedad de base , asi com evitar la precipitación de las Crioglobulinas para evitar su efecto obstructivo e inflamatorio.
Los AINES se pueden usar en pacientes con artralgias y fatiga, y los inmunosupresores(corticoides, ciclofosfamida o azatioprina) están indicados cuando hay evidencia de compromiso orgánico tal como vasculitis, enfermedad renal progresiva, trastornos neurológicos o manifestaciones dérmicas severas. La plasmaféresis se indica en complicaciones que ponen en peligro la vida como crioprecipitación o hiperviscosidad. El PEG Interferon alfa combinado con Rivabirina ha demostrado eficacia en pacientes con Crioglobulinemia asociada a Hepatitis C. Pequeños ensayos con Rituximab (Ac monoclonal quimérico anti CD20), ha mostrado efectividad
Nuestra paciente tuvo una mejoría espontánea en el seguimiento. No se llevó a cabo biopsia hepática por tratarse de una paciente de mas de 70 años. La actividad inflamatoria hepática ha permanecido silente desde el punto de vista de laboratorio, y mejoraron concomitantemente las lesiones de piel asi com el compromiso renal. Nunca se vió afectada la función renal y el clearence de creatinina fue siempre normal para la edad. El sedimento de orina se normalizó y la única manifestación que ha persistido en el tiempo luego de 4 años de seguimiento es la neuropatía periférica
ARTRITIS INTERMITENTE
ARTRITIS INTERMITENTE
Paciente de 26 años, sexo femenino, que consultó por haber presentado artritis de rodilla.
Antecedentes de hipotiroidismo en tto con T4, 1 embarazo con parto normal. Sin antecedentes familiares de importancia.
EA: la paciente refiere que desde hace 8 años sufre cuadros de artritis franca de una u otra rodilla, con marcado dolor y notable tumefacción y rubor, que duran aproximadamente 3 días acompañado de rigidez matinal y que ceden generalmente con AINES IM que le dan en la guardia del Hospital Municipal de Tandil. Estos cuadros se repetían en dos oportunidades por año, pero últimamente los brotes se repiten cada 3 meses aprox. En forma esporádica presenta lumbalgia.
Al exámen, llama la atención la facies con labios afinados y nariz afilada, con leve exoftalmos.
No presentaba artritis al exámen físico. No Raynaud, sin lesiones en piel. No existen anormalidades al exámen respiratorio.
Se solicitó rx de Fergusson, de rodillas y de columna dorsolumbar que fueron normales.
LABO: HTO 39, BL 8700, CON FÓRMULA CONSERVADA, FAN-, ERS 86 mm 1h, LATEX-, PCR-, Proteinograma normal. Orina completa normal.
Cuando trajo éstos resultados refiere haber presentado otro cuadro de artritis de rodilla, de comienzo nocturno, acompañado de rigidez al levantarse. Cedió al 2do día con AINES IM.
El exámen físico tampoco mostró esta vez artritis, aunque se notaba inestabilidad articular de rodillas.
Se solicitaron HIV, y Huddleson que fueron -. ERS 72.
Con diagnóstico presuntivo de Reumatismo Palindrómico se inició tto con Hidroxicloroquina 400 mg/día.
Se solicitó RMN de ambas rodillas.
Gustavo Rabazzano
Antecedentes de hipotiroidismo en tto con T4, 1 embarazo con parto normal. Sin antecedentes familiares de importancia.
EA: la paciente refiere que desde hace 8 años sufre cuadros de artritis franca de una u otra rodilla, con marcado dolor y notable tumefacción y rubor, que duran aproximadamente 3 días acompañado de rigidez matinal y que ceden generalmente con AINES IM que le dan en la guardia del Hospital Municipal de Tandil. Estos cuadros se repetían en dos oportunidades por año, pero últimamente los brotes se repiten cada 3 meses aprox. En forma esporádica presenta lumbalgia.
Al exámen, llama la atención la facies con labios afinados y nariz afilada, con leve exoftalmos.
No presentaba artritis al exámen físico. No Raynaud, sin lesiones en piel. No existen anormalidades al exámen respiratorio.
Se solicitó rx de Fergusson, de rodillas y de columna dorsolumbar que fueron normales.
LABO: HTO 39, BL 8700, CON FÓRMULA CONSERVADA, FAN-, ERS 86 mm 1h, LATEX-, PCR-, Proteinograma normal. Orina completa normal.
Cuando trajo éstos resultados refiere haber presentado otro cuadro de artritis de rodilla, de comienzo nocturno, acompañado de rigidez al levantarse. Cedió al 2do día con AINES IM.
El exámen físico tampoco mostró esta vez artritis, aunque se notaba inestabilidad articular de rodillas.
Se solicitaron HIV, y Huddleson que fueron -. ERS 72.
Con diagnóstico presuntivo de Reumatismo Palindrómico se inició tto con Hidroxicloroquina 400 mg/día.
Se solicitó RMN de ambas rodillas.
Gustavo Rabazzano
Suscribirse a:
Entradas (Atom)