La mejor manera de evitar diagnósticos erróneos, las principales características clínico-patológicas e inmunológicas y las guías prácticas respecto de las terapias.Autor/a: Dalakas MC. Inflammatory Muscle Diseases N Engl J Med 2015; 372:1734-47 DOI: 10.1056/NEJMra1402225
Introducción
Las miopatías inflamatorias son el mayor grupo de miopatías potencialmente tratables en niños y adultos. Constituyen un grupo heterogéneo de trastornos que están mejor clasificados, en función de las características clínico-patológicas distintas, en cuatro subtipos: dermatomiositis, polimiositis, miositis necrotizante autoinmune, y miositis por cuerpos de inclusión (a lo largo de esta revisión, se utiliza este término para referirse específicamente a la miositis esporádica por cuerpos de inclusión). Un quinto subtipo, llamado miositis de superposición, también está empezando a ser reconocido.
Es fundamental la identificación del subtipo correcto y la distinción de estas condiciones a partir de otras enfermedades que tienen características que imitan a estas condiciones, porque cada subtipo tiene un pronóstico y la respuesta a las terapias es diferente. Esta revisión refleja el conocimiento actual de estas condiciones, destaca la mejor manera de evitar diagnósticos erróneos, describe las principales características clínico-patológicas e inmunológicas, y proporciona guías prácticas respecto a las terapias.
Características clínicas generales
Los pacientes con miopatías inflamatorias tienen dificultad creciente con las tareas que requieren el uso de los músculos proximales, como levantarse de una silla, subir escaleras o tareas que implican levantar objetos. Las tareas que requieren el uso de músculos distales, como abotonarse o sostener objetos, son afectadas a principios de la miositis por cuerpos de inclusión, pero sólo en casos avanzados de polimiositis, dermatomiositis y miositis necrotizante autoinmune.
Los músculos oculares se libran en todos los subtipos, pero los músculos faciales están comúnmente afectados en la miositis por cuerpos de inclusión en todos los subtipos de la enfermedad, los músculos extensores del cuello y los faríngeos pueden participar, lo que se traduce en dificultades para sostener la cabeza (caída de la cabeza) o en disfagia. En casos agudos avanzados y raros, pueden verse afectados los músculos respiratorios.
La atrofia muscular se detecta temprano en la miositis por cuerpos de inclusión, con atrofia selectiva de los cuádriceps y los músculos del antebrazo, pero se desarrolla en todos los subtipos si la debilidad es severa y crónica. Se puede producir sensibilidad muscular y mialgia, especialmente en pacientes con el síndrome de antisintetasa (ver el glosario), pero si el dolor es intenso y la debilidad sigue un patrón "separatista", en el que el paciente tiene dificultad para mantener el esfuerzo, debe ser descartada la fascitis o la fibromialgia.
En todas las miopatías inflamatorias pueden ocurrir manifestaciones extramusculares, a pesar de que se producen en la miositis por cuerpos de inclusión sólo en casos excepcionales; estas manifestaciones incluyen síntomas sistémicos, como fiebre, artralgia, y el fenómeno de Raynaud, como se ve en los sindromes antisintetasa, arritmias cardíacas o disfunción ventricular, en casos relativamente raros en la que el músculo cardíaco afectado es clínicamente sintomático; y las complicaciones pulmonares, debido principalmente a la enfermedad pulmonar intersticial, que se presentan en 10 a 40% de pacientes.
La prevalencia de la enfermedad pulmonar intersticial, una condición que se detecta mejor con tomografía computada de alta resolución, es tan alta como 70% en los pacientes con anti-histidil-ARN transferasa (ARNt) sintetasa (anti-Jo-1) o la proteína asociada a la diferenciación anti-melanoma (MDA) -5 anticuerpos (ver el Glosario).
Características clínicas específicas
Dermatomiositis
Las características clínicas específicas de las miopatías inflamatorias se describen en la Tabla 1. La dermatomiositis se ve en niños y adultos, y los primeros síntomas incluyen manifestaciones cutáneas distintas que acompañan o son anteriores a la debilidad muscular; las manifestaciones cutáneas incluyen heliotropo periorbital (azul-violeta) exantema con edema; erupción eritematosa en la cara, las rodillas, los codos, maléolos, cuello, tórax anterior (en signo de V), y en la espalda y los hombros (signo del chal); y una erupción violácea (erupción de Gottron) sobre los nudillos, que puede evolucionar en una escala de decoloración.
Las lesiones son fotosensibles y se pueden agravar por radiación ultravioleta, capilares dilatados bucles en la base de la uñas, cutículas irregulares y engrosadas y dedos palmares agrietados ("manos de mecánico") son características de las calcificaciones subcutáneas de la dermatomiositis.
Las calcificaciones subcutáneas, a veces se extruyen a la superficie de la piel y pueden ocurrir ulceraciones e infecciones, y son especialmente comunes entre los niños. Si la resistencia del paciente parece ser normal, la dermatomiositis puede limitarse a la piel (dermatomiositis amiopática), a pesar de que la afectación muscular subclínica es frecuente. En los niños, un síntoma temprano es "la desdicha", definida como la irritabilidad combinada con un rubor en la cara, fatiga, y una renuencia a socializarse.
Los síntomas de la dermatomiositis puede superponerse con los de la esclerosis sistémica y enfermedades mixtas del tejido conectivo; en tales casos, la erupción típica de la piel es transitoria o leve. La miositis de superposición está empezando a ser reconocida como una entidad distinta; se manifiesta sin la erupción que es típica de la dermatomiositis, con prominentes cambios patológicos en las regiones perifascicular, interfascicular y perimisial, y se asocia con frecuencia con anticuerpos antisintetasa.
En los adultos, el riesgo de cáncer se incrementa durante los primeros 3 a 5 años después del inicio de la dermatomiositis, con una frecuencia reportada de 9 a 32% .Los cánceres más comunes son el cáncer de ovario, cáncer de mama, cáncer de colon, melanoma, cáncer de la nasofaringe (en los asiáticos), y el linfoma no Hodgkin; el riesgo de estos cánceres requiere un chequeo anual profundo en los primeros 3 años después del comienzo de la enfermedad.
Polimiositis
La polimiositis es rara como una entidad independiente y con frecuencia se diagnostica erróneamente; la mayoría de los pacientes cuya condición ha sido diagnosticada como polimiositis suelen tener miositis por cuerpos de inclusión, miositis necrotizante autoinmune o distrofia inflamatoria. La polimiositis sigue siendo un diagnóstico de exclusión y se define mejor como una miopatía proximal subaguda en adultos que no tienen erupción, historia familiar de enfermedad neuromuscular, exposición a drogas miotóxicas (por ejemplo, estatinas, penicilamina y zidovudina), participación de los músculos faciales y extraoculares, endocrinopatía, o el fenotipo clínico de miositis por cuerpos de inclusión.
Miositis necrotizante autoinmune
La miositis necrotizante autoinmune es una entidad clínico patológica distinta que se produce con más frecuencia que la polimiositis, y representa hasta un 19% de todas las miopatías inflamatorias. Puede ocurrir a cualquier edad, pero es vista principalmente en los adultos; se inicia ya sea de forma aguda, alcanzando su pico durante un período de días o semanas, o subaguda, progresando de manera constante y causa debilidad grave y muy altos niveles de creatinkinasa.
La miositis necrotizante autoinmune se produce sola o después de infecciones virales, en asociación con cáncer, en pacientes con trastornos del tejido conectivo como la esclerodermia, o en pacientes que toman estatinas, en los que la miopatía sigue empeorando después de la suspensión de las estatinas (si la miopatía mejora dentro de 4 a 6 semanas después de la interrupción de las estatinas, se debe probablemente a los efectos tóxicos del fármaco en lugar de a la miopatía inmune).
La mayoría de los pacientes con miositis autoinmunes necrotizantes tienen anticuerpos contra la partícula de reconocimiento de señal (SRP) o agonistas de 3-hidroxi-3-metilglutaril-coenzima A reductasa (HMGCR) (ver el Glosario).
Miositis por cuerpos de inclusión
La miositis por cuerpos de inclusión es la miopatía inflamatoria más común e incapacitante entre las personas de 50 años de edad o mayores. Su prevalencia, que se estimó inicialmente en los Países Bajos como 4,9 casos por millón de habitantes, es mucho mayor cuando se ajusta por edad; en dos estudios posteriores en Australia y Estados Unidos, la prevalencia ajustada por edad varió desde 51,3 hasta 70 casos por millón. En un pequeño estudio de revisión gráfica realizado en un condado de Estados Unidos, la incidencia estimada de la miositis por cuerpos de inclusión fue de 7,9 casos por millón en las décadas de 1980 y 1990.
La enfermedad comienza de forma insidiosa y se desarrolla durante un período de años, a veces de forma asimétrica (es decir, puede iniciar o ser más grave en un extremo o en un lado del cuerpo), y progresa de forma constante, simulando una distrofia muscular en la vejez o una enfermedad de la motoneurona lentamente progresiva.
Aunque la miositis por cuerpos de inclusión se sospecha comúnmente cuando un paciente con presunta polimiositis no responde a la terapia, 3 características que pueden conducir a un diagnóstico clínico temprano incluyen:
- Participación temprana de los músculos distales, especialmente los extensores y flexores de los dedos del pie.
- Aatrofia de los músculos de los antebrazos y cuádriceps; caídas frecuentes debido a la debilidad de los músculos cuádriceps causando pandeo de las rodillas.
- Debilidad muscular facial leve.
Los músculos axiales pueden verse afectados, lo que resulta en camptocormia (flexión hacia adelante de la columna vertebral) o caída de la cabeza. La disfagia se produce en más del 50% de los pacientes.
Diagnóstico
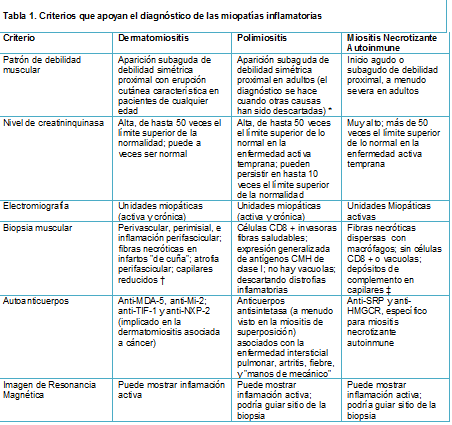
El diagnóstico del subtipo exacto de miopatía inflamatoria se basa en la combinación de la historia clínica, el tiempo de la progresión de la enfermedad, el patrón de afectación muscular, los niveles de enzimas musculares, los hallazgos electromiográficos, el análisis de la biopsia muscular, y para algunas condiciones, la presencia de ciertos autoanticuerpos (Tabla 1).
Los cambios típicos en la piel, con o sin debilidad muscular, indican dermatomiositis; un inicio subagudo de puntos proximales de debilidad miopáticos a polimiositis o miositis autoinmune necrotizante; y debilidad lentamente progresiva proximal y distal con puntos selectivos de atrofia a la miositis por cuerpos de inclusión. La electromiografía es útil para el diagnóstico en todos los subtipos de la enfermedad para descartar condiciones neurogénicas y evaluar la actividad de la enfermedad.
La actividad en suero de la creatinkinasa está elevada en todos los subtipos, pero niveles muy altos desde el punto de inicio sugiere miositis necrotizante autoinmune.
Las imágenes de resonancia magnética (RMI) son útiles para el diagnóstico cuando está presente el edema muscular o se sospecha miofascitis, así como para la identificación de los músculos particulares afectados por la atrofia en la miositis por cuerpos de inclusión.
La biopsia muscular es esencial para el diagnóstico de polimiositis, miositis de superposición, miositis necrotizante autoinmune, y miositis por cuerpos de inclusión, así como para descartar patologías que imitan la enfermedad tales como distrofias, miopatías metabólicas o vacuolares.
La evaluación de autoanticuerpos es útil para el diagnóstico de miositis autoinmune necrotizante y para la clasificación de los distintos subtipos y sus asociaciones con la participación de órganos sistémicos, como la enfermedad pulmonar intersticial.
Entre las enzimas derivadas del músculo en el suero, el indicador más sensible de miopatía inflamatoria es la creatinkinasa, que está elevada en los pacientes con enfermedad activa. Los niveles más altos, de hasta más de 50 veces el límite superior de la normalidad, se observan en pacientes con miositis autoinmune necrotizante, y los más bajos (menos de 10 veces el límite superior de la normalidad) se observan en pacientes con miositis por cuerpos de inclusión.
Aunque los niveles en suero de creatinkinasa por lo general son paralelos a la actividad de la enfermedad, pueden ser normales o sólo ligeramente elevados en pacientes con dermatomiositis activa, miositis de superposición, o miositis activa por cuerpos de inclusión.
Junto con la creatinkinasa, también son elevados los niveles de aminotransferasa y aspartato alanina aminotransferasa, un signo que a veces se interpreta erróneamente como indicio de enfermedad hepática y conduce a una investigación con una biopsia de hígado en lugar de una biopsia muscular. Los niveles de aldolasa en suero pueden ser también elevados, especialmente si la fascia está involucrada.
La electromiografía puede mostrar los potenciales de la unidad motora miopática (de corta duración, unidades polifásicas de baja amplitud sobre la activación voluntaria) y aumento de la actividad espontánea con fibrilaciones, descargas repetitivas complejas y ondas agudas positivas. Estos hallazgos son útiles para determinar si la miopatía es activa o crónica y para descartar trastornos neurogénicos, pero no pueden ser utilizados para diferenciar miopatías inflamatorias de miopatías distróficas o tóxicas.
Las imágenes de RNM pueden utilizarse para identificar edema, inflamación en el músculo o la fascia, infiltración grasa, fibrosis o atrofia. Son útiles para evaluar el alcance y la selectividad de la afectación muscular, especialmente en los casos de miositis por cuerpos de inclusión; para la identificación de actividad de la enfermedad; y para guiar la selección del músculo con el mayor grado de inflamación para biopsiar.
El examen de muestras de biopsia de músculo revela características distintas a cada subtipo de la enfermedad, y aunque los resultados no siempre son típicos o específicos, sigue siendo la herramienta de diagnóstico más importante. La biopsia muscular es más útil cuando el sitio de la biopsia se elige adecuadamente (es decir, en un músculo que no tiene signos clínicos de enfermedad avanzada o terminal, pero tampoco mínimamente afectado), la muestra se procesa en un laboratorio con experiencia, y la hallazgos son interpretados en el contexto de la clínica.
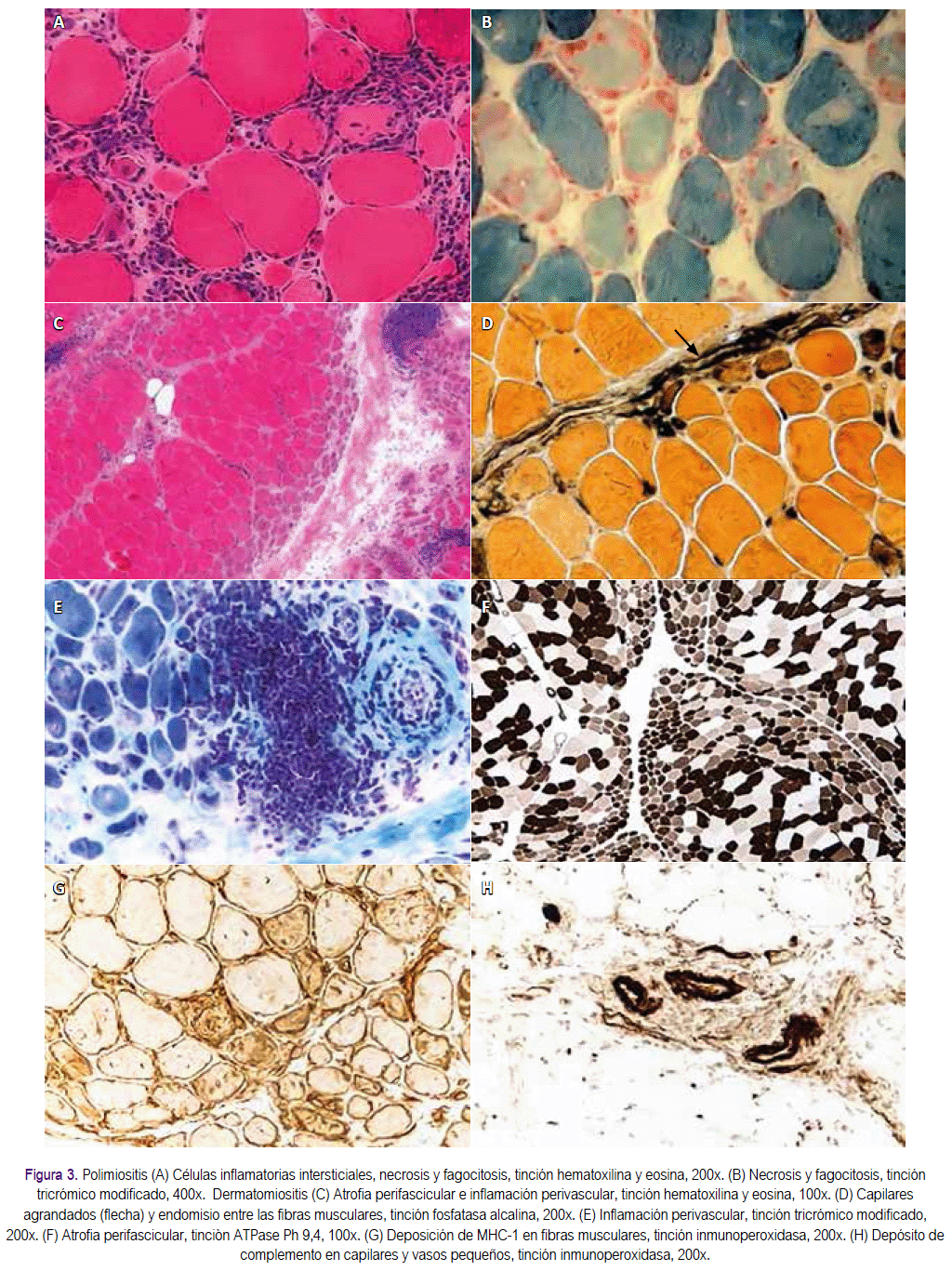
En la dermatomiositis, la inflamación es perivascular y está más prominentemente situada en los septos interfasciculares o en la periferia de los fascículos. Las fibras musculares sufren necrosis y fagocitosis - a menudo en una porción de un fascículo muscular o de la periferia del fascículo - debido a micro infartos que conducen a hipoperfusión y atrofia perifascicular. La atrofia perifascicular, que se caracteriza por capas de fibras atróficas en la periferia de los fascículos, a menudo con infiltrados perivasculares e interfasciculares, es diagnóstico de dermatomiositis (o de miositis de superposición, cuando los cambios en la piel están ausentes o son transitorios).
En la polimiositis y miositis por cuerpos de inclusión, la inflamación es perivascular y se concentra más típicamente en focos múltiples dentro del endomisio; compuesta fundamentalmente por células T CD8 + invasoras de aspecto saludable, las fibras musculares no necróticas expresan el antígeno del complejo mayor de histocompatibilidad (CMH) de clase I (las fibras musculares normales no expresan este antígeno). El hallazgo de la expresión del CMH y las células T CD8 + (denominado complejo CMH-CD8) es útil para confirmar el diagnóstico y para descartar trastornos con inflamación no inmune, como se ve en algunas distrofias musculares.
En la miositis necrotizante autoinmune hay abundantes fibras necróticas invadidas o rodeadas por macrófagos. Los infiltrados linfocitarios son escasos, y la regulación del CMH clase 1 es a menudo prominente entre las fibras necróticas.
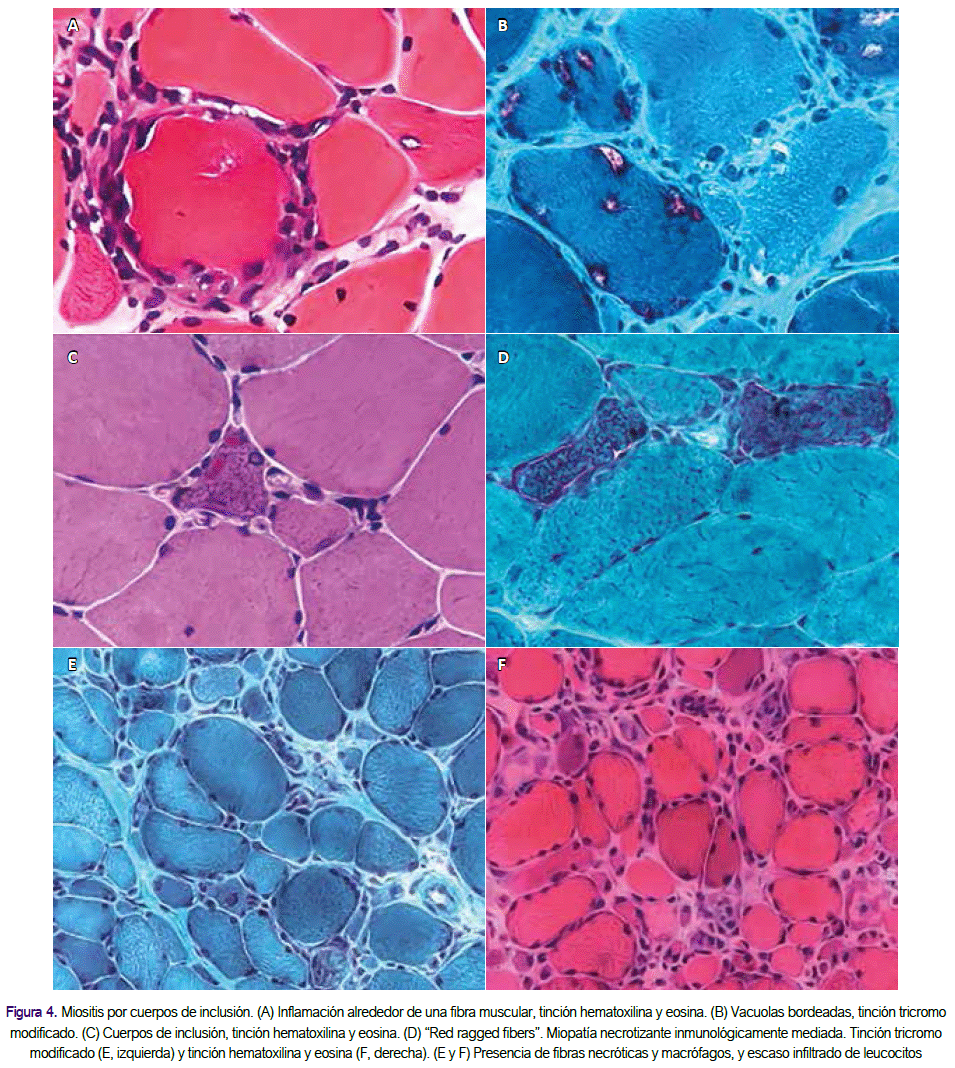
La miositis necrotizante autoinmune es más a menudo mediada por anticuerpos específicos contra SRP o HMGCR (ver el Glosario), a menudo con depósitos de complemento en los capilares. La miositis por cuerpos de inclusión tiene todas las características inflamatorias de la polimiositis, incluyendo el complejo CD8-CMH, pero además tiene cambios miopáticos crónicos con incrementos en el tejido conectivo y en la variabilidad de tamaño de la fibra, vacuolas autofágicas que tienen paredes forradas internamente con un material que se tiñe rojo azulado con hematoxilina y eosina o tricrómico de Gomori modificado, "rojo-irregular” o fibras citocromo oxidasa negativo al que representan las mitocondrias anormales y depósitos amiloides congofílicos junto a las vacuolas, que son mejor visualizados con cristal violeta o fluorescente óptico.
La microscopía electrónica muestra filamentos tubulares de 12 a 16 nm de diámetro junto a las vacuolas. En hasta el 30% de los pacientes con el fenotipo clínico típico de miositis por cuerpos de inclusión, no se encuentran vacuolas o depósitos de amiloide en la muestra de biopsia muscular y sólo es vista la inflamación, lo que conduce a un diagnóstico erróneo de polimiositis. Tales pacientes tienen "miositis por cuerpos de inclusión clínica" diagnosticada en base a criterios de correlación clínico-patológicos basados en datos que confirman debilidad en las fibras del dedo flexor o del cuádriceps, inflamación alrededor de las fibras no necróticas con la expresión de CMH de clase I, y citocromo oxidasa-negativos, incluso sin vacuolas, que son específicos para el diagnóstico de miositis por cuerpos de inclusión clínica.
Los autoanticuerpos dirigidos contra ARN nucleares o antígenos citoplasmáticos se detectan en hasta el 60% de los pacientes con miopatías inflamatorias, dependiendo de la serie de casos y el método de detección utilizado. Aunque el papel patogénico de los anticuerpos no está claro, algunos parecen ser específicos para diferentes fenotipos clínicos y genotipos HLA-DR. Estos anticuerpos incluyen aquellos contra sintetasa aminoacil tRNA (ARS), que se detectan en 20 a 30% de los pacientes.
Entre los ocho ARS diferentes que han sido identificados, anti-Jo-1, el anticuerpo más ampliamente disponible comercialmente, representa el 75% de todas las antisintetasas asociadas con el síndrome de antisintetasa. Este síndrome se caracteriza por la miositis con destacados cambios patológicos en la periferia de los fascículos y el tejido conectivo perimisial, enfermedad pulmonar intersticial, artritis, fenómeno de Raynaud, fiebre, y manos de mecánico. En un caso poco frecuente se encontraron células T γ δ para reconocer ARS, que representaron el primer eslabón patogénico entre ARS e inmunidad mediada por células.
Los anticuerpos específicos de miositis necrotizantes autoinmunes se dirigen contra la traslación de la proteína de transporte SPR o contra HMGCR, el objetivo farmacológico de las estatinas.
Anti-HMGCR, visto en el 22% de las personas con miositis necrotizante autoinmune, independientemente del uso de estatinas, se correlaciona con los niveles de creatinkinasa y la fuerza. La dermatomiositis asociada a anticuerpos incluyen anti-Mi-2, que está asociado con las lesiones típicas de la piel; anti-MDA-5, que se asocia principalmente con la dermatomiositis amiopática o enfermedad pulmonar intersticial; y anti-factor de transcripción 1γ intermediario (anti-TIF-1γ) y proteína anti-nuclear de la matriz 2 (anti-NXP-2), que suelen estar presentes en pacientes adultos con dermatomiositis asociada a cáncer, aunque su presencia está influenciada por factores geográficos, raciales y genéticos.
Se detecta anti-citosólica de 5'-nucleotidasa 1A (anti-CN1A) en 60 a 70% de los pacientes con miositis por cuerpos de inclusión, aunque el grado de sensibilidad y especificidad varía de acuerdo con el método de detección utilizado, e indica la activación de células B.
Mecanismos Patológicos
Inmunopatología
Las causas de las miopatías inflamatorias son desconocidas, pero una patogenia autoinmune está fuertemente implicada. En la dermatomiositis, el complejo del complemento C5b-9 de ataque membranolítico se activa temprano (es evidente antes de la destrucción de las fibras musculares) y se deposita sobre las células endoteliales, lo que lleva a la necrosis, la reducción de la densidad de los capilares endomisiales, isquemia y destrucción de la fibra muscular que asemeja microinfartos, los capilares dilatados restantes tienen lúmenes para compensar la isquemia.
La atrofia perifascicular residual refleja la hipoperfusión endofascicular, que es más prominente en la periferia de los fascículos. La activación del complejo de ataque de membrana, presumiblemente por anticuerpos, desencadena la liberación de citoquinas proinflamatorias, hasta regula las moléculas de adhesión en las células endoteliales, y facilita la migración de los linfocitos activados, incluyendo las células B, células T CD4 +, y células dendríticas plasmocitoides, a los espacios perimisial y endomisial.
La inmunidad innata desempeña también un papel que se basa en el aumento de la expresión de proteínas inducibles por interferón tipo I en la región perifascicular, una zona donde también se sobre expresan otras moléculas inflamatorias, degenerativas, o regenerativas; y queda por determinar si el efecto de la inmunidad innata es causado por el gen inducible por ácido retinoico 1 de señalización en respuesta a las señales locales de las fibras dañadas, lo que conduce a autoamplificación de la inflamación perifascicular mediante la activación de interferón-β y el CMH de clase I. En la dermatomiositis juvenil, las células quiméricas maternas pueden contribuir a la patogénesis de la enfermedad.
En la polimiositis y la miositis por cuerpos de inclusión, las células T CD8 + citotóxicos rodean e invaden a las fibras musculares de aspecto saludable, no necróticas que expresan el CMH clase I aberrante. La expresión del CMH de clase I, que está ausente en el sarcolema de las fibras musculares normales, es probablemente inducida por citoquinas secretadas por células T activadas. El complejo CD8-MHC de clase I es característico de la polimiositis y miositis por cuerpos de inclusión, y su detección ayuda a la confirmación del diagnóstico histológico.
Las células T CD8+ contienen gránulos de perforina dirigidos hacia la superficie de las fibras musculares, que causan mionecrosis sobre la liberación. El análisis de moléculas del receptor de células T expresado por las células T CD8+ infiltrantes revela la expansión clonal de las cadenas de células T del receptor y las secuencias conservadas en la región de unión al antígeno, lo que sugiere una respuesta de células T impulsadas por antígeno. Esto se ve apoyado por la expresión de moléculas co estimuladoras y de regulación de moléculas de adhesión, quimiocinas, y citokinas.
Las células Th17 y T reguladoras participan en el proceso inmunológico. La regulación y la sobrecarga de CMH de clase I también pueden causar mal plegamiento de glicoproteínas, que hace hincapié en el retículo endoplásmico de las fibras musculares. La activación de células B también ocurre, más prominente en la miositis por cuerpos de inclusión (aunque no está claro si el músculo puede sostener formaciones l de centro germinal), en la que también se detectan autoanticuerpos anti-CN1A (ver el Glosario).
Los factores que desencadenan enfermedades musculares inflamatorias siguen siendo desconocidos. Han sido propuestos factores de riesgo genéticos que regulan la respuesta inmune contra agentes ambientales indefinidos. Las interacciones genéticas son apoyados por las asociaciones entre HLA-DRB1 * 03 y anti-Jo-1, entre el HLA-DRB1 * 11: 01 y entre anti-HMGCR positivo y miositis necrotizante autoinmune, y entre HLA-DRB1 * 03: 01 y HLA-DRB1 * 01: 01 y con miositis por cuerpos de inclusión.
Los virus pueden ser responsables de la interrupción de la tolerancia inmune, pero los intentos para amplificar virus- incluyendo virus coxsackie, virus influenza, paramixovirus (incluyendo el virus de la papera), citomegalovirus y virus EpsteinBarr- desde los músculos tienen fallas. La mejor evidencia para una conexión viral se refiere a un retrovirus, porque la polimiositis o miositis por cuerpos de inclusión se desarrolla en las personas infectadas con el virus de la inmunodeficiencia humana (VIH) o virus linfotrópico de células T humana I. Sin embargo, los antígenos retrovirales se detectaron sólo en los macrófagos del endomisio y no dentro de las fibras musculares.
Las células T auto invasivas están impulsadaspor clonación, y algunas están asociadas a retrovirus específicos. La polimiositis asociada al VIH o la miositis por cuerpos de inclusión asociada a VIH, deben distinguirse de una miopatía mitocondrial tóxica inducida por medicamentos antirretrovirales, que mejora cuando los medicamentos son discontinuados.
Componente degenerativo de la miositis por cuerpos de inclusión
La miositis por cuerpos de inclusión es un trastorno complejo debido a que, además del componente de autoinmunidad, hay un importante componente degenerativo, destacado por la presencia de depósitos de amiloide congofílico dentro de algunas fibras. Similar a lo que se ve en la enfermedad de Alzheimer, estos depósitos inmunorreaccionan contra la proteína amiloide precursora, amiloide-β42, apolipoproteína E, α-sinucleína, presenilina, ubiquitina, y tau fosforilada, que indican la presencia de la agregación de proteínas.
Depósitos de TDP43, una proteína de unión aberrante al ADN, trasladadas desde los núcleos al citoplasma, y p62, una proteína de enlace que transporta ubiquitinas, detectadas dentro de las fibras musculares con el uso de la inmunotinción, se han defendido como pruebas marcadoras de diagnóstico. La evidencia in vitro sugiere que el amiloide β42 y su oligómero están involucrados en la vía de la toxicidad intracelular, pero no queda claro cómo estos agregados proteicos, que también se ven en otras miopatías vacuolares, inducen una miopatía inflamatoria y degenerativa y lo que desencadena la enfermedad, la inflamación, o el agregado de proteínas.
La microdisección láser de fibras de células T invadidas, en comparación con fibras no invadidas o vacuoladas ha revelado la regulación diferencial sobre la regulación de la señalización inflamatoria, como la señalización del receptor del interferón γ.
La evidencia sugiere que el envejecimiento, la proteostasis anormal (red de control de las proteínas), la alteración de la autofagia, el estrés celular inducido por el CMH de clase I o de óxido nítrico, la inflamación de larga data, y las citocinas proinflamatorias como el interferón-γ y la interleucina-1β57, pueden desencadenar o acumulativamente disparar la degeneración, lo que lleva a una mayor acumulación de moléculas estresantes y proteínas mal plegadas.
Tratamiento de Dermatomiositis, Polimiositis y Miositis Necrotizante autoinmune
La prednisona oral administrada una vez al día después del desayuno en una dosis de 1 mg por kilo de peso corporal, hasta 100 mg por día, es el fármaco de primera línea para el tratamiento de la dermatomiositis, polimiositis y miositis necrotizante autoinmune; esta elección de medicamentos se basa en la experiencia, pero no en estudios controlados. Algunos médicos prefieren añadir un agente inmunosupresor desde el principio.
En los pacientes en que la enfermedad empeora rápidamente, es preferible administrar metilprednisolona intravenosa a una dosis de 1000 mg por día durante 3 a 5 días antes de comenzar el tratamiento con glucocorticoides orales. Después de 3 a 4 semanas, la prednisona se disminuye, según lo dictado por la respuesta de la enfermedad a la terapia, preferiblemente para ir de una dosis diaria a dosis en días alternos, sin embargo, si los signos objetivos de aumento de la fuerza y la capacidad para realizar actividades de la vida diaria están ausentes en ese momento, se acelera la disminución de modo que se pueda iniciar el tratamiento con el siguiente agente disponible.
Un error táctico es la práctica de "perseguir" el nivel de creatinkinasa como una señal de respuesta, especialmente en pacientes que reportan una sensación de sentirse mejor, pero no necesariamente de sentirse más fuertes. Cuando la fuerza aumenta, el nivel de creatinkinasa sérica cae, pero una disminución sola de la creatinkinasa no es un signo de mejoría.
Para pacientes en los que los glucocorticoides producen una respuesta, azatioprina, mofetil micofenolato, metotrexato o ciclosporina pueden ser utilizados empíricamente para ahorrar glucocorticoides. Cuando co-existe enfermedad pulmonar intersticial, pueden ser útiles la ciclofosfamida o el tacrolimus.
En pacientes con dermatomiositis, se recomiendan los glucocorticoides tópicos o inhibidores de la calcineurina y evitar la luz solar. Cuando fallan los glucocorticoides para inducir la remisión o en casos severos y rápidamente progresivos, es apropiada la terapia de inmunoglobulina intravenosa (2 g por kilogramo en dosis divididas durante un período de 2 a 5 días consecutivos).
En un estudio doble ciego, se encontró que la inmunoglobulina intravenosa era eficaz en el tratamiento de la dermatomiositis refractaria; pueden ser necesarias infusiones mensuales para mantener la remisión. En ensayos abiertos, la inmunoglobulina intravenosa también ha parecido ser eficaz en el tratamiento de la polimiositis y la miositis necrotizante autoinmune. La inmunoglobulina subcutánea parece que mantiene la remisión en estudios no controlados, a pequeña escala.
Si la enfermedad no ha respondido a los glucocorticoides y la inmunoglobulina intravenosa, el paciente debe ser reevaluado, y si hay incertidumbres diagnósticas, debe considerarse repetir una biopsia muscular. Si se volvió a confirmar el diagnóstico, se han probado agentes biológicos que han sido aprobados para el tratamiento de otras enfermedades inmunes y pueden considerarse como opción de tratamiento experimental. Estos incluyen rituximab (un anticuerpo anti-CD20), que a una dosis de 2 g (dividido en dos infusiones con 2 semanas de diferencia) parece eficaz en algunos pacientes con dermatomiositis, polimiositis, miositis necrotizante o autoinmune.
En un estudio controlado con placebo con 200 pacientes, en la semana 8 no hubo diferencia entre el grupo placebo y el grupo de rituximab, y sobre la base del diseño del estudio, los resultados no fueron significativos; sin embargo, en la semana 44, cuando todos los pacientes habían recibido rituximab, el 83% respondió a la definición de mejoría. Los pacientes con anticuerpos anti-SRP anti-Jo-1, Antimi-2, o parecen más propensos a tener una respuesta.
Los inhibidores del factor de necrosis tumoral (infliximab, adalimumab, etanercept) son ineficaces y pueden empeorar o desencadenar la enfermedad. Otros productos biológicos que pueden ser considerados como tratamiento experimental incluyen alemtuzumab, que al parecer es eficaz en polimiositis; anti-complemento C3 (eculizumab), que es eficaz en enfermedades mediadas por complemento y puede ser eficaz para el tratamiento de la dermatomiositis y miositis necrotizante autoinmune; anti-interleucina-6 (tocilizumab) y anti-receptor de la interleucina (anakinra), que han sido eficaces en casos anecdóticos; anti-interleucina-17; y anti-interleuquina-1β (gevokizumab), que se está evaluando en un ensayo en curso (número EudraCT, 2012-005772-34). En general, el resultado a largo plazo de las miopatías inflamatorias ha mejorado sustancialmente, con una tasa de supervivencia a los 10 años de más del 90%.
Tratamiento de Miositis por cuerpos de Inclusión
Debido a los efectos citotóxicos mediados por las células T y la mejora relacionada con amiloides en proteínas agregadas por citoquinas proinflamatorias en pacientes con miositis por cuerpos de inclusión, han sido probados agentes inmunosupresores como tratamiento para este subtipo de enfermedad, pero todos han fracasado, probablemente debido a que la enfermedad comienza mucho antes de que los pacientes consulten al médico, cuando la cascada degenerativa ya está avanzada.
Los glucocorticoides, metotrexato, ciclosporina, azatioprina y micofenolato son ineficaces, y aunque algunos pacientes pueden tener inicialmente niveles de mejoría subjetivos leves al ser tratados con uno de estos agentes, no se logra ningún beneficio a largo plazo. La inmunoglobulina intravenosa se ha considerado eficaz en ensayos controlados de forma transitoria, pero puede ayudar a algunos pacientes, especialmente aquellos con disfagia. Alemtuzumab puede proporcionar estabilización a corto plazo, pero se necesita un estudio controlado.
El tratamiento con anakinra tampoco ha sido exitoso. Los ensayos dirigidos a moléculas de TGF-β-musculares o factores de crecimiento muscular están en progreso. Bimagrumab, un anticuerpo que inhibe la señalización de un receptor superfamilia de TGF-β, se demostró en un estudio a pequeña escala que aumenta el volumen muscular después de 8 semanas, lo que ha llevado a un estudio controlado en curso (número ClinicalTrials.gov, NCT01925209). Se ha completado un pequeño estudio controlado, prueba de concepto de arimoclomol (número ClinicalTrials.gov, NCT00769860), un agente que hasta regula la respuesta de las proteínas de choque térmico y atenúa el estrés celular; la droga tenía un perfil de efectos adversos aceptable, pero si hubo beneficios clínicamente significativos sigue siendo desconocido.
En la actualidad, los tratamientos sintomáticos son la mejor opción. Para la disfagia potencialmente mortal que no responde a la inmunoglobulina intravenosa, puede ser considerada la dilatación o miotomía del cricofaríngeo. Al igual que con todas las miopatías inflamatorias, ejercicios de resistencia no fatigantes y terapias ocupacionales y de rehabilitación son útiles para mejorar la deambulación, evitar caídas, evitar la atrofia por desuso, y prevenir contracturas conjuntas. Aunque la esperanza de vida de los pacientes con miositis por cuerpos de inclusión es, en la mayoría de los pacientes normal, en la etapa final de la enfermedad, requieren dispositivos de asistencia, como un bastón, andador o silla de ruedas.
Glosario
- Anti–cytosolic 5′-nucleotidase 1A (anti-cN1A, or anti-NT5C1A): Autoanticuerpos dirigidos contra la proteína nuclear CN1A implicado en el procesamiento del ARN; asociado con miositis por cuerpos de inclusión.
- Anti–histidyl–transfer RNA synthetase (anti-Jo-1): El autoanticuerpo más común asociado con el síndrome de antisintetasa, que consiste en miopatía, fiebre, enfermedad pulmonar intersticial, fenómeno de Raynaud, artritis, y "manos de mecánico."
- Anti–3-hydroxy-3-methylglutaryl–coenzyme A reductase (anti-HMGCR): Autoanticuerpos dirigidos contra HMGCR, la diana farmacológica de las estatinas; específico para miositis necrotizante autoinmune.
- Anti–melanoma differentiation–associated protein-5 (anti-MDA-5): Autoanticuerpos dirigidos contra una helicasa específica de ARN citoplásmico; asociados con dermatomiositis amiopática o enfermedad pulmonar intersticial rápidamente progresiva.
- Anti-Mi-2: Autoanticuerpos dirigidos contra una helicasa de ADN nuclear implicada en la activación transcripcional; asociados con lesiones de la piel típicas de la dermatomiositis.
- Anti–signal recognition particle (anti-SRP): Autoanticuerpos dirigidos contra un complejo polipéptidico implicado en el transporte de proteínas al retículo endoplásmico; específico para miositis necrotizante autoinmune.
- Anti–transcriptional intermediary factor 1 γ (anti-TIF-1γ): Autoanticuerpos implicados en el crecimiento y la diferenciación celular; visto en la dermatomiositis asociada a cáncer, junto con proteína de la matriz nuclear anti-2 (anti-NXP-2).
COMENTARIO
Esta revisión describe claramente las principales características de las miopatías inflamatorias. Constituyen un grupo heterogéneo de enfermedades potencialmente tratables, que tienen características clínico patológicas diferentes, en las que se basa su clasificación. Hay 4 subtipos descriptos: polimiositis, dermatomiositis, miositis por cuerpos de inclusión y miositis necrotizante autoinmune y un quinto, que está empezando a ser reconocido.
La identificación del subtipo y el diagnóstico diferencial con otras patologías que tienen algunas características en común, es fundamental para enfocar el tratamiento adecuado al más corto plazo. Esto es importante porque cada subtipo tiene tratamiento específico y diferente respuesta a la terapéutica implementada.