Autor/a: Dres. Annelies Berden, Arda Göçeroğlu1, David Jayne, Raashid Luqmani, Niels Rasmussen, Jan Anthonie Bruijn, Ingeborg Bajema BMJ 2012;344:e26
Las vasculitis asociadas con anticuerpos contra el citoplasma de los neutrófilos (ANCA) son las enfermedades autoinmunes sistémicas de causa desconocida que afectan a los vasos sanguíneos de tamaño pequeño y mediano.
Estas enfermedades son:
- Granulomatosis con poliangeítis (anteriormente Granulomatosis de Wegener).
- Poliangeítis microscópica
- Poliangeítis granulomatosa eosinofílica (anteriormente Sme. de Churg-Strauss).
Esta revisión se centra principalmente en la granulomatosis con poliangeitis y la poliangeítis microscópica. A pesar de que son relativamente poco frecuentes, deben ser diagnosticadas y tratadas a tiempo, porque la enfermedad no tratada puede desarrollar rápidamente un fallo multiorgánico y la muerte. Con los tratamientos modernos, estas enfermedades no son fatales, pero sí crónicas.
El diagnóstico precoz y el tratamiento pueden prevenir la progresión al daño en los órganos y alargar la vida más sana. Un estudio reciente de pacientes con vasculitis asociada a ANCA halló un retraso de 3 a 12 meses entre la aparición de la enfermedad y el diagnóstico, lo que sugiere que el retraso diagnóstico es un problema.
¿Quiénes las sufren?
La incidencia anual de vasculitis asociada a ANCA en Europa y América del Norte es de aproximadamente 20 por millón (en el Reino Unido en 2.008, la prevalencia puntual para la granulomatosis con poliangeítis era de 130/millón y para la poliangeítis microscópica, de 47.9/millón). El inicio de la enfermedad generalmente se presenta entre los 65 y los 74 años, aunque puede ocurrir a cualquier edad.
En general, la prevalencia es más elevada en los hombres, pero las mujeres suelen desarrollar la enfermedad a una edad joven. La prevalencia global de la vasculitis asociada a ANCA es mayor en las personas de raza blanca. La incidencia de la granulomatosis con poliangeitis es mayor en el norte de Europa, mientras que la de la poliangeítis microscópica es mayor en el sur de Europa y Japón.
¿Cómo se presentan los pacientes?
Los pacientes típicamente se presentan con síntomas prodrómicos de "influenza", de varias semanas o meses de duración, como fiebre, polimialgia, poliartralgias, cefalea, malestar general, anorexia y pérdida de peso involuntaria. Estos síntomas inespecíficos se superponen a los síntomas de procesos no vasculíticos tales como el síndrome posviral, las infecciones o los tumores malignos.
Se debe considerar el diagnóstico diferencial de vasculitis en los pacientes con síntomas generales y signos de enfermedad inflamatoria. Algunos pacientes inicialmente pueden presentar la enfermedad vasculitis focal, con erupción cutánea, rinitis hematicopurulenta, escleritis o artritis. En tales pacientes, el examen cuidadoso de otros órganos y sistemas puede mostrar otras manifestaciones de la enfermedad.
Los pacientes pueden presentar síntomas diferentes a través del tiempo. Los síntomas de las diferentes vasculitis asociadas a ANCA se superponen, pero algunos de los síntomas son más comunes en ciertas enfermedades. Por ejemplo, pueden presentarse problemas del oído, la nariz y la garganta - pérdida de la audición, otalgia, (con sangre), rinorrea, otorrea, sinusitis, formación de costras nasales y otitis media recurrente, como ocurre en aproximadamente el 90% de los pacientes con granulomatosis con poliangeítis y en el 35% de las personas con poliangeítis microscópica.
Un gran estudio de observación ha demostrado que las vías aéreas y el parénquima pulmonar son comúnmente afectados, como son los riñones, aunque esto puede no ser evidente hasta que ocurre la insuficiencia renal. Por lo tanto, el análisis de orina puede identificar la afectación renal al comienzo de la enfermedad. Casi el 50% de los pacientes tiene manifestaciones cutáneas de la enfermedad como urticaria o nódulos cutáneos sensibles. También son comunes la afectación de los ojos y del sistema nervioso. Es necesario un examen cuidadoso para establecer la extensión de la enfermedad.
¿Cómo se puede diagnosticar?
Investigaciones que se pueden realizarse en atención primaria
En los análisis de sangre solicitados en atención primaria puede hallarse leucocitosis, trombocitosis, velocidad de eritrosedimentación y proteína C reactiva elevadas, anemia normocítica normocrómica, creatinina sérica elevada. Los pacientes con síntomas y signos de vasculitis y alteraciones en estos exámenes de sangre requieren un análisis de orina, incluyendo el examen del sedimento urinario, para buscar hematuria y proteinuria. Un aumento de la creatininemia indica que hay daño renal. La radiografía de tórax en los pacientes con síntomas respiratorios como disnea, tos o hemoptisis puede mostrar infiltrados, nódulos, o cavidades en el parénquima pulmonar.
En atención primaria se puede solicitar un análisis de ANCA, y está indicado ante un paciente que presenta una enfermedad inexplicable que ha durado más de un par de semanas y se asocia con una velocidad de eritrosedimentación o proteína C reactiva elevadas, en particular, si tiene más de un sistema orgánico afectado. Cuatro grandes ensayos controlados aleatorizados internacionales comprobaron que, antes del tratamiento, la prueba es positiva en el 90-95% de los pacientes con granulomatosis con poliangeítis activa generalizada o poliangeítis microscópica. Los dos tipos de análisis que se utilizan generalmente son: la inmunofluorescencia indirecta (IFI) y el ensayo por inmunoabsorción ligado a enzima (ELISA).

Un estudio de observación multicéntrico internacional halló que la IFI es más sensible pero que ELISA es más especifico. La norma estándar internacional actual es el uso de la IFI como prueba de detección y del ELISA como prueba de confirmación de los resultados positivos. El análisis de ANCA se debe realizar solo en laboratorios con experiencia. Las pruebas no son estándar, de manera que la sensibilidad y la especificidad varían entre laboratorios y los valores de referencia no están disponibles.
Aunque el análisis de ANCA es positivo en la mayoría de los pacientes con enfermedad no tratada, un resultado negativo no excluye el diagnóstico de vasculitis asociada a ANCA porque el 5-10% de los pacientes no desarrollan estos anticuerpos. Tampoco una prueba negativa de ANCA excluye la presencia de otros síndromes de vasculíticos de los vasos pequeños y medianos no asociados a ANCA. Tales pacientes pueden requerir una investigación más sistemática para determinar el grado de su enfermedad.
Aunque en la atención secundaria la prueba de ANCA es ampliamente utilizada como una herramienta de detección de rutina para la vasculitis, en este grupo de pacientes tiene poca sensibilidad y especificidad. Cuando hay una indicación más clara y probabilidad de vasculitis, el rendimiento de la prueba es mayor. Si hay un elevado índice de sospecha sobre la base de los hallazgos clínicos, pero la prueba de ANCA es negativa, es conveniente repetirla un par de semanas después, pero si los pacientes tienen manifestaciones de la enfermedad en varios sitios, deben ser derivados inmediatamente al especialista.
El paciente con un resultado positivo en la prueba de ANCA será derivado al especialista que corresponda, según las manifestaciones de la enfermedad (reumatólogo, nefrólogo o posiblemente un neumonólogo o un cirujano de nariz, garganta y oído).

La derivación a especialistas sin experiencia en vasculitis puede retrasar el diagnóstico. Muchas afecciones pueden estar asociadas a un resultado positivo en la prueba de ANCA, incluyendo la enfermedad inflamatoria intestinal, las infecciones crónicas (como la tuberculosis) y las enfermedades autoinmunes como el lupus eritematoso sistémico y la artritis reumatoidea, mientras que los ANCA también pueden ser inducidos por diversos fármacos. Esto pone de relieve la necesidad de pruebas prudentes.
Para interpretar los resultados, es fundamental tener en cuenta el contexto clínico en el que se realiza la prueba. Los autores recomiendan el análisis de ANCA de forma rutinaria en las siguientes circunstancias: enfermedad aguda o crónica destructiva de vías aéreas superiores; evidencia de enfermedad inflamatoria renal, como lo indica un sedimento urinario activo o los análisis bioquímicos indicativos de progresión rápida los parámetros de glomerulonefritis, evidencia de enfermedad pulmonar inflamatoria (como lo indican los síntomas clínicos o las alteraciones radiológicas), vasculitis cutánea asociada con enfermedad sistémica, y la mononeuritis múltiple.
El análisis de ANCA también se indica en la estenosis subglótica de la tráquea, la cual se manifiesta como disnea lentamente progresiva, y en presencia de una masa retroorbitaria con protrusión del bulbo ocular y diplopía, aunque sin pruebas especializadas estas condiciones pueden ser difíciles de reconocer.
Estudios realizados en atención primaria
La tomografía computarizada proveerá información adicional sobre la ubicación y naturaleza de las lesiones identificadas en la radiografía de tórax. El diagnóstico de vasculitis asociada a ANCA se confirma por las anormalidades específicas halladas en las biopsias de tejido obtenidas de los sitios de enfermedad activa -vasculitis, células gigantes, necrosis “geográfica” y granulomas.
En la granulomatosis con poliangeítis, las biopsias de las vías respiratorias, principalmente la nariz y los senos paranasales, a menudo no muestran más de uno de los signos histopatológicos característicos. En estos casos, el médico tiene que tratar al paciente sobre la base de los resultados clínicos típicos o una prueba positiva de ANCA (o ambos). Varias biopsias de lesiones activas, si es posible tomadas de diferentes órganos en momentos diferentes, aumentan la posibilidad de establecer un diagnóstico histológico.
Cuando hay afectación renal, las biopsias renales tienen una mayor rendimiento diagnóstico; por lo general, muestran una cantidad variable de glomerulonefritis focal necrotizante. Un estudio internacional aleatorizado con control ciego investigó más de 95 biopsias renales y pudo establecer el diagnóstico histopatológico de glomerulonefritis asociada a ANCA en el 80-98% de las biopsias.
¿Cuál es el curso natural de la enfermedad si no se trata?
Un estudio clave de 1958 sobre la historia natural de la enfermedad en 56 pacientes halló que la supervivencia promedio fue de unos 5 meses, el 82% de los pacientes no sobrevivió al primer año después del diagnóstico y más del 90% de los pacientes murió dentro de los 2 años. La principal causa de muerte fue "Uremia" como resultado de la insuficiencia renal rápidamente progresiva, y la segunda causa más común fue la insuficiencia respiratoria.
¿Cuál es el tratamiento actual de la vasculitis?
El tratamiento estándar consiste en inducir la remisión con una dosis elevada de glucocorticoides y pulsos con dosis elevadas de ciclofosfamida oral o intravenosa durante 3-6 meses; el mantenimiento de la remisión se hace con azatioprina o metotrexato mientras que los glucocorticoides se van reduciendo lentamente hasta su suspensión.
Como tratamiento de inducción, los ensayos han utilizado un régimen de pulsos de ciclofosfamida intravenosa durante un mínimo de 6 meses. Los cursos de pulsos de ciclofosfamida intravenosa de menos de 6 meses pueden seguir siendo apropiados cuando se logra el control rápido de la enfermedad.
El tratamiento requiere la supervisión de un especialista con el objeto controlar la actividad de la enfermedad, para evitar mayores daños a los órganos y prevenir la recurrencia de las vasculitis. El manejo de la toxicidad del tratamiento es una parte importante del cuidado del paciente y el médico general puede enfrentarse a este problema De acuerdo con un gran estudio aleatorizado no ciego, el 75% de los pacientes tratados con ciclofosfamida oral diaria y prednisolona logró la remisión durante 3 meses. En ese momento, la prednisolona generalmente se reduce a 10-15 mg/día, mientras que la ciclofosfamida se suspende por el riesgo de toxicidad acumulativa siendo reemplazada por un inmunosupresor alternativo. Los pacientes que no responden inicialmente al tratamiento de inducción deben continuar durante más tiempo con el tratamiento pudiendo considerarse los tratamientos de segunda línea.
Se desconoce cuál es la duración óptima del tratamiento de mantenimiento y la práctica difiere ampliamente entre los centros asistenciales. Los agentes inmunosupresores de mantenimiento alternativos, como el micofenolato micofenolato, podrían estar indicados en casos seleccionados. Aunque para la enfermedad limitada o menos grave se ha utilizado el metotrexato como terapia de inducción en lugar de la ciclofosfamida, potencialmente más tóxica, un ensayo controlado aleatorizado no ciego de 100 pacientes comprobó que se asociaba a una tasa de recurrencia mayor, por lo que su uso sigue siendo controvertido.
A largo plazo, se requieren visitas regulares al especialista (cada 3 meses por lo menos) para comprobar la actividad de la enfermedad, los efectos secundarios del tratamiento y manejar las consecuencias de los daños irreversibles en los tejidos, como la insuficiencia renal. Con los modernos tratamientos, la vasculitis asociada a ANCA ha pasado de ser una enfermedad con amenaza inminente a la vida a ser una enfermedad crónica propensa a las recaídas durante toda la vida. Un gran estudio de observación de 107 pacientes encontró que alrededor del 50% de los pacientes tratados experimentó 1 o más recaídas durante 5 años.
Los estudios de seguimiento a largo plazo han demostrado claramente que la reducción de la exposición a la ciclofosfamida se asocia con un mayor riesgo de recaída tardía, por lo que se necesita un equilibrio entre la reducción de la exposición a la ciclofosfamida y el aumento del riesgo de recaída. Los datos recogidos prospectivamente de 524 pacientes mostraron que los riesgos importantes del tratamiento con ciclofosfamida incluyen la infección, la infertilidad, y el cáncer incidental. El tratamiento a largo plazo con corticosteroides también tiene muchos efectos secundarios. Estos efectos adversos impulsan la búsqueda de tratamientos más eficaces y seguros.
Nuevos agentes terapéuticos en estudio
En dos ensayos controlados aleatorizados prospectivos recientes se comprobó que el agente rituximab, reductor de las células B, es eficaz para inducir la remisión en los pacientes con vasculitis asociada a ANCA, y que su perfil de seguridad es comparable al del tratamiento estándar. Es necesaria investigación adicional sobre la eficacia y la seguridad de este fármaco, a pesar de que fue aprobado recientemente por la Food and Drug Administration de EE.UU. para ser usado en combinación con fármacos para tratar a pacientes con granulomatosis con poliangeítis y poliangeítis microscópica.
Para los pacientes con enfermedad renal grave y como complemento del tratamiento estándar, se ha investigado el intercambio de plasma. En 2007, un gran ensayo controlado aleatorizado internacional de 137 pacientes halló mejores resultados con el intercambio de plasma que con la metilprednisolona como tratamiento coadyuvante, con respecto a la recuperación de la función renal. Sin embargo, un metaanálisis reciente concluyó que se necesitan más datos para establecer el beneficio a largo plazo de este tratamiento.
En general, el intercambio de plasma es seguro. Un gran estudio de 7.538 intercambios en 887 pacientes mostró efectos secundarios y técnicos incidentes en el 16,8% de todos los intercambios. Esto incluyó una reacción transfusional en el 6,9%, una tasa de flujo insuficiente en el 5%, hipotensión en el 2,9%, alteraciones electrocardiográficas en el 1,8%, hipocalcemia en el 1,4%, colapso en el 0,9% e insuficiencia pulmonar en el 0,5%. El intercambio de plasma se interrumpió solo en el 4% de los casos.
¿Cuál es la perspectiva a largo plazo para los pacientes con vasculitis asociada a ANCA?
Con el tratamiento moderno, la enfermedad ha pasado de ser universalmente mortal a ser una enfermedad crónica con recaídas y remisiones. A menudo se afectan varios órganos pero el compromiso renal es un resultado habitual, apareciendo glomerulonefritis en la etapa renal terminal con insuficiencia y necesidad de reemplazo renal en el 20-40% de los pacientes (resultados de a estudios de observación con una mediana de seguimiento de 3,1 a ≥ 5 años).
El riesgo de muerte para los pacientes tratados con los regímenes terapéuticos actuales sigue siendo 2,6 veces superior al de los controles comparados por edad. El mayor riesgo de muerte predomina en el primer año después del diagnóstico, cuando las infecciones y la vasculitis activa son las causas de la mayoría de las muertes prematuras. Los pacientes mayores con insuficiencia renal grave tienen un riesgo particularmente elevado de morir a los pocos meses de la presentación, lo que refleja la gravedad de su enfermedad, como así su mayor susceptibilidad a la toxicidad de los tratamientos actuales.
La mortalidad de los pacientes que sobreviven al primer año después del diagnóstico sigue siendo 1,3 veces mayor que la de los controles de la población a edades comparativas. La muerte después del primer año se debe principalmente a las infecciones, las enfermedades cardiovasculares y el cáncer.
Los pacientes están en riesgo permanente de infecciones y, a menudo es necesario el tratamiento con antibióticos. En una estudio de control de casos de base poblacional, muchos pacientes dijeron tener fatiga como uno de los problemas principales, lo cual afectaba su empleo y en general su calidad de vida. El impacto socioeconómico de la enfermedad, sin embargo, ha resultado difícil de evaluar.
¿Qué deben tener en cuenta los médicos generalistas con respecto al tratamiento?
Los pacientes pueden recurrir a su médico de cabecera para recibir apoyo e información. Las primeras semanas de tratamiento pueden ser difíciles porque los pacientes, por lo general, continúan teniendo los síntomas asociados a la vasculitis. Se necesitan visitas al hospital y análisis de sangre frecuentes, con el fin de monitorear la actividad de la enfermedad y la respuesta al tratamiento. Antes de cada tratamiento con dosis elevadas de ciclofosfamida, hay que tener la precaución de que el recuento de plaquetas, leucocitos, especialmente el neutrófilos, y el hepatograma tengan valores estables. La dosis debe ajustarse de acuerdo al nivel de creatinina en sangre.
La ciclofosfamida oral diaria, la azatioprina, o el micofenolato mofetil y el metotrexato requieren un el seguimiento semanal de rutina con hemograma, hepatograma, exámenes de la función renal y función hepática, para evitar la toxicidad de los medicamentos. Por lo general, para estos controles de rutina, a los pacientes es les es más conveniente asistir al consultorio de atención primaria, pero esto requiere una buena comunicación entre la atención primaria y secundaria. Las recomendaciones sobre cómo se debe modificar el tratamiento en respuesta a los resultados inesperados deben ser acordados antes del tratamiento.
Los pacientes son susceptibles a las infecciones, en particular durante el tratamiento de inducción (en su mayoría, de las vías respiratorias y urinarias), siendo necesaria la intervención temprana con antibióticos de las infecciones confirmadas. En el caso de los pulsos de ciclofosfamida, que habitualmente se administran con antieméticos y antimicóticos orales, el momento más vulnerable para los pacientes es a los 7-10 días después de cada pulso. El medicamento suprime la médula ósea, causa neutropenia y aumenta el riesgo de infección. Se aconseja que los pacientes no entren en contacto con personas con infecciones de las vías respiratorias bajas o superiores durante este tiempo.
Los pacientes tratados con pulsos de ciclofosfamida pueden llegar a estar profundamente cansados en los 2-3 días después de la administración, pero mejorarán poco a poco. Sin embargo, hay que tener en cuenta las posibles interacciones medicamentosas. Por ejemplo, los pacientes con granulomatosis con poliangeítis pueden ser tratados con dosis elevadas de metotrexato y desarrollar neutropenia grave si una infección del tracto urinario es tratada con la dosis estándar de cotrimoxazol; por lo tanto, como tratamiento estándar concomitante del metotrexato se sugiere la profilaxis con dosis bajas cotrimoxazol (960 mg, 3 veces/semana).
Recordar que deben considerarse las consecuencias de los daños causados por la enfermedad, como la enfermedad renal crónica, y los efectos del tratamiento, como la osteoporosis y el control glucémico por la intolerancia a la glucosa causada por la exposición crónica a los glucocorticoides. Los pacientes también tienen mayor riesgo de cáncer y aterosclerosis prematura y acelerada, lo que les predispone a enfermedades cardiovasculares, incluyendo el accidente cerebrovascular. Es importante controlar y tratar todos los factores de riesgo cardiovascular bien conocidos.
Tres estudios de observación encontraron una mayor incidencia de eventos tromboembólicos venosos, que no parecen ser atribuibles a los factores de riesgo protromótico clásicos. Para prevenir el daño a la mucosa gástrica provocado por las altas dosis de glucocorticoides se indican los inhibidores de la bomba de protones. Los pacientes que toman inmunosupresores suelen requerir profilaxis con cotrimoxazol, debido a un mayor riesgo de adquirir neumonía por Pneumocystis jiroveci.
Debido a que la recaída puede ocurrir después de muchos años de remisión, los pacientes suelen permanecer en seguimiento indefinido. Los brotes de la enfermedad son comunes y los pacientes deben ser alentados a buscar atención médica urgente cuando experimentan una reactivación es decir, recurrencia, deterioro o una nueva aparición de los síntomas y signos atribuibles a la vasculitis activa.
Si los pacientes tienen una erupción o experimentan complicaciones graves del tratamiento puede ser necesaria la hospitalización. Si se omiten o ignoran los primeros síntomas puede sobrevenir un episodio grave de vasculitis con insuficiencia renal o respiratoria. Los pacientes deben recibir instrucciones para reconocer los primeros síntomas de recaída y actuar en consecuencia, ya que esta mala evolución puede ser prevenida.

¿Cuáles son las causas de vasculitis?
Aunque las causas exactas se desconocen, la vasculitis asociada a ANCA probablemente sea el resultado de una interacción entre factores genéticos y ambientales. Se han hecho varias asociaciones con los genes que codifican a las proteínas implicadas en la inmunidad y que también se asocian a menudo con otras enfermedades autoinmunes. Sin embargo, los miembros de la familia o los gemelos de los pacientes con granulomatosis con poliangeítis rara vez manifiestan la enfermedad, lo que va en contra de un fuerte componente de predisposición genética.
Un reciente estudio sueco con registros del país mostró mayor incidencia familiar de la granulomatosis con poliangeítis, lo que podría denotar una susceptibilidad genética a la enfermedad, pero también puede ser que hayan stado expuestos a los mismos factores ambientales capaces de inducir el efecto dentro de un grupo familiar. Entre los factores ambientales implicados se mencionan la exposición ocupacional al sílice (como la agricultura, el trabajo en la construcción), los antitiroideos y los fármacos antihipertensivos (propiltiouracilo e hidralazina), y varios agentes microbianos, especialmente S. aureus.
El catarro nasal crónico de este fármaco se ha asociado con una mayor incidencia de recaída en la granulomatosis con poliangeítis, y un estudio multicéntrico aleatorizado, doble ciego demostró que el tratamiento profiláctico con cotrimoxazol reduce la incidencia de recaídas de la granulomatosis con poliangitis.
Todos estos factores pueden jugar un papel en el desarrollo de los ANCA, que generalmente son considerados participantes de la patogénesis de la enfermedad. La mayor parte de la evidencia clínica directa de su intervención es el desarrollo de un síndrome renopulmonar en un recién nacido poco después de haber nacido de una madre con poliangeítis microscópica mieloperoxidasa (MPO)-ANCA positiva, probablemente debido a la transmisión transplacentaria de la madre MPO-ANCA.
Sin embargo, no se han reportado otros casos, mientras que un niño a término normal y saludable acaba de nacer de una madre con poliangeítis microscópica, a pesar de la transferencia transplacentaria de MPO-ANCA. El desarrollo de un modelo de ratón en el que la inyección de glomerulonefritis inducida por MPO-ANCA y vasculitis, similar a la observada en la enfermedad humana, ofrece mucha evidencia in vivo para la patogenicidad de estos anticuerpos. Sin embargo, hasta la fecha no se ha desarrollado un modelo de la misma calidad para la proteinasa 3 (PR3)-ANCA. En la vasculitis asociada a ANCA ha quedado firmemente establecido el papel fundamental de los neutrófilos en la lesión aguda de la pared de los vasos sanguíneos.
Se cree que la base de la acumulación local de neutrófilos en la enfermedad se debe a la imprimación de los neutrófilos circulantes por las citocinas, posiblemente durante la infección. La imprimación de los neutrófilos provoca la expresión de PR3 y MPO por su membrana celular, donde se vuelven accesibles al ANCA. Los neutrófilos activados por ANCA se desgranulan, producen especies reactivas de oxígeno y liberan enzimas proteolíticas que dañan las paredes de los vasos sanguíneos.
Aparte de los ANCA y los neutrófilos, es probable que también sean de importancia las alteraciones de la inmunidad celular. En los últimos años también se ha propuesto como importante la vía alternativa del complemento. Debido a que la glomerulonefritis asociada a ANCA en el ser humano es pauci-inmune, con prácticamente ninguna deposición del complemento en el tejido renal, se ha asumido que el complementan no representa ningún papel en esta enfermedad. Sin embargo, estudios experimentales recientes apoyan este papel en la vasculitis pauci-inmune asociada a MOP-ANCA.

♦ Traducción y resumen objetivo: Dra. Marta Papponetti. Especialista en Medicina Interna.

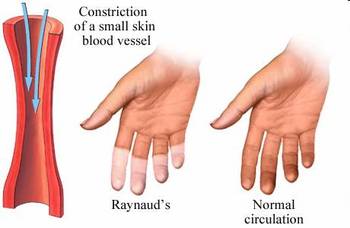 El examen debe tener en cuenta los antecedentes personales. En las manos buscar cambios de color, cambios en el lecho ungular y en la integridad de la piel. La esclerodactilia, las deformidades en flexión, las fricciones tendinosas y la calcinosis se ven en la esclerosis sistémica. Las úlceras digitales no son normales y siempre reflejan el fenómeno de Raynaud secundario; estas úlceras deben motivar enseguida el examen de otros signos de una enfermedad del tejido conectivo y la derivación al especialista.
El examen debe tener en cuenta los antecedentes personales. En las manos buscar cambios de color, cambios en el lecho ungular y en la integridad de la piel. La esclerodactilia, las deformidades en flexión, las fricciones tendinosas y la calcinosis se ven en la esclerosis sistémica. Las úlceras digitales no son normales y siempre reflejan el fenómeno de Raynaud secundario; estas úlceras deben motivar enseguida el examen de otros signos de una enfermedad del tejido conectivo y la derivación al especialista.