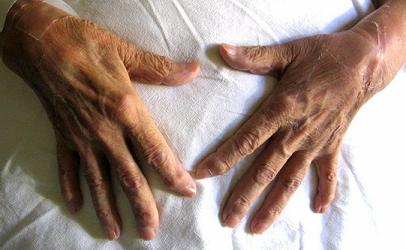
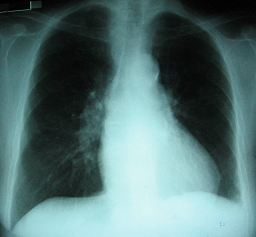
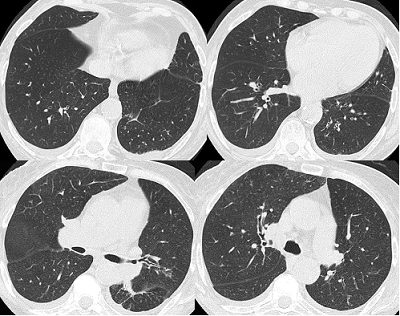
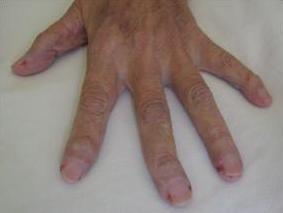
Paciente femenina de 51 años con poliartritis febril, adenomegalias hiliares y uveítis.Sarcoidosis.
Paciente femenina de 51 años 

Motivo de consulta: poliartritis febril.
Antecedentes personales y familiares:
Migraña crónica.
Síndrome depresivo menor, actualmente en remisión sin medicación.
Hipertrigliceridemia.
Hipotiroidismo.
Padre fallecido de cáncer de pulmón. Hermanos 2, ambos fallecidos uno por tumor cerebral a los 70 años, madre fallecida por “leucemia” a los 42 años, y otro por ruptura de aneurisma inflamatorio de ao rta abdominal por Salmonella tiphy.Hijos 2 vivos y sanos.
rta abdominal por Salmonella tiphy.Hijos 2 vivos y sanos.
Enfermedad actual: Paciente oriunda de Azul, que en pleno estado de salud comienza en Agosto de 2009 a sentir dolores generalizados de tipo gripales, seguido al cabo de unos días de artritis de tobillo derecho, al que se agrega rodilla homolateral al cabo de 24 hs. Posteriormente, en el término de 4 días agrega artritis de metacarpofalángicas, y hombro derecho. Los dolores de dichas articulaciones le impiden realizar sus actividades. La poliartritis tenía características de migratriz. El cuadro presentaba además registros subfebriles que no superaban los 38ºC durante casi todo el día con escasa variación circadiana aumentando levemente a la tarde.
El resto del examen físico era normal.
No existía sudoración nocturna, cambios de coloración de la orina, escalofríos, rash cutáneo,conjuntivitis, piodermitis ni forunculosis, flujo ginecológico, síntomas urinarios bajos, ni alteraciones del tránsito intestinal. No había mayor compromiso del estado general, con apetito conservado.
Al cabo de 1 semana se agrega tos seca, y disnea de esfuerzo clase funcional 2/3, y dolor ocular con instalación de “ojo rojo” derecho.
Se solicitan análisis de laboratorio: Gr 4350000 Hb 11,6, Hto 38%. Gb 9500 granulocitos 70%. Linfocitos 22% eosinófilos 1%. ESD 42 mm. Hepatograma normal.
Hemocultivos negativos. Urocultivo negativo.
Latex AR negativo. FAN (+) 1/80. ASTO 73. Ferritina plasmática normal. Serología para Hepatitis A,B y C negativas. CMV negativos Epstein-Barr negativo. Parvovirus y Rubeola negativos. PPD negativa.
Dosaje de enzima convertidora de angiotensina 57,4 U/L (normal 8 a 52)
Proteinograma electroforético normal.
Se le realizó una Rx de tórax que mostró dudosos hilios adenopáticos bilaterales por lo que se solicitó una TAC tóraco-abdómino-pélvica.
Se solicitó examen ginecológico que fue normal no considerándose necesario la toma de muestras para examen de gonococo, Chlamydia, micoplasma.
TAC de tórax:
Presencia de adenomegalias axilares izquierdas. Mediastino adenomegalias pretraqueales, en ventana aórtico-pulmonar, subcarinales, pre-aórticas y en ambos hilios pulmonares. Se observa la presencia de micronódulos en ambas bases pulmonares.
ECG: taquicardia sinusal.
Se realizó un eco-cardiograma que mostró función ventricular izquierda normal con cavidades de tamaño normal, y pequeño derrame pericárdico leve a moderado.
Se realizó interconsulta a oftalmología quien interpretó el cuadro ocular como uveítis anterior.
Se comenzó tratamiento con AINES (Naproxeno 500 mg/12 hs) con lo que se obtuvo mejoría parcial.
La paciente siguió refiriendo dolores articulares y agregó malestar general, anorexia, y continuó por 1 semana con registros subfebriles. Intensificó la tos seca y la disnea que se hizo aún para esfuerzos menores.
Cuál es el diagnóstico?

Migraña crónica.
Síndrome depresivo menor, actualmente en remisión sin medicación.
Hipertrigliceridemia.
Hipotiroidismo.
Padre fallecido de cáncer de pulmón. Hermanos 2, ambos fallecidos uno por tumor cerebral a los 70 años, madre fallecida por “leucemia” a los 42 años, y otro por ruptura de aneurisma inflamatorio de ao
 rta abdominal por Salmonella tiphy.Hijos 2 vivos y sanos.
rta abdominal por Salmonella tiphy.Hijos 2 vivos y sanos.Enfermedad actual: Paciente oriunda de Azul, que en pleno estado de salud comienza en Agosto de 2009 a sentir dolores generalizados de tipo gripales, seguido al cabo de unos días de artritis de tobillo derecho, al que se agrega rodilla homolateral al cabo de 24 hs. Posteriormente, en el término de 4 días agrega artritis de metacarpofalángicas, y hombro derecho. Los dolores de dichas articulaciones le impiden realizar sus actividades. La poliartritis tenía características de migratriz. El cuadro presentaba además registros subfebriles que no superaban los 38ºC durante casi todo el día con escasa variación circadiana aumentando levemente a la tarde.
El resto del examen físico era normal.
No existía sudoración nocturna, cambios de coloración de la orina, escalofríos, rash cutáneo,conjuntivitis, piodermitis ni forunculosis, flujo ginecológico, síntomas urinarios bajos, ni alteraciones del tránsito intestinal. No había mayor compromiso del estado general, con apetito conservado.
Al cabo de 1 semana se agrega tos seca, y disnea de esfuerzo clase funcional 2/3, y dolor ocular con instalación de “ojo rojo” derecho.
Se solicitan análisis de laboratorio: Gr 4350000 Hb 11,6, Hto 38%. Gb 9500 granulocitos 70%. Linfocitos 22% eosinófilos 1%. ESD 42 mm. Hepatograma normal.
Hemocultivos negativos. Urocultivo negativo.
Latex AR negativo. FAN (+) 1/80. ASTO 73. Ferritina plasmática normal. Serología para Hepatitis A,B y C negativas. CMV negativos Epstein-Barr negativo. Parvovirus y Rubeola negativos. PPD negativa.

Dosaje de enzima convertidora de angiotensina 57,4 U/L (normal 8 a 52)
Proteinograma electroforético normal.
Se le realizó una Rx de tórax que mostró dudosos hilios adenopáticos bilaterales por lo que se solicitó una TAC tóraco-abdómino-pélvica.
Se solicitó examen ginecológico que fue normal no considerándose necesario la toma de muestras para examen de gonococo, Chlamydia, micoplasma.
TAC de tórax:
Presencia de adenomegalias axilares izquierdas. Mediastino adenomegalias pretraqueales, en ventana aórtico-pulmonar, subcarinales, pre-aórticas y en ambos hilios pulmonares. Se observa la presencia de micronódulos en ambas bases pulmonares.
ECG: taquicardia sinusal.
Se realizó un eco-cardiograma que mostró función ventricular izquierda normal con cavidades de tamaño normal, y pequeño derrame pericárdico leve a moderado.

Se realizó interconsulta a oftalmología quien interpretó el cuadro ocular como uveítis anterior.
Se comenzó tratamiento con AINES (Naproxeno 500 mg/12 hs) con lo que se obtuvo mejoría parcial.
La paciente siguió refiriendo dolores articulares y agregó malestar general, anorexia, y continuó por 1 semana con registros subfebriles. Intensificó la tos seca y la disnea que se hizo aún para esfuerzos menores.
Cuál es el diagnóstico?
La paciente fue derivada al Hospital Italiano de Buenos Aires con diagnóstico presuntivo de sarcoidosis para realizar estudios anatomopatológicos de adenomegalias mediastinales.
En dicho establecimiento se realizaron los siguientes estudios:
Examen funcional respiratorio sin hallazgos patológicos en la espirometría.
DLCO (difusión de CO), normal.

Barrido corporal con Galio 67:
Se realizó barrido corporal total, con adquisición de imágenes planares a las 24 y 48 hs y tomográficas (SPECT) de tórax a las 48 hs.
Se observa un aumento de captación del trazador en proyección de ambos hilios pulmonares (signo de Lambda) y en proyección de ambas glándulas lagrimales y fosas nasales.
Las imágenes descriptas se corroboran con la adquisición Spect de tórax.
Los patrones centelleográficos descriptos son compatibles con el diagnóstico de sarcoidosis en actividad .

Se realizó mediastinoscopía con toma de muestras de adenomegalias mediastinales. El informe preliminar del profesional que llevó a cabo la biopsia por congelación del ganglio obtenido por mediastinoscopía fue de “granulomas no caseosos compatibles con sarcoidosis”.
Se comienza tratamiento con:
Deltisona 40 mg/día.
Carbonato de calcio 1 comp por día.
Raquiferol 7 gotas por semana.
Omeprazol 20 mg/ día.
Clonacepan 0,5 mg/día.
A las 48 horas de comenzado el tratamiento mejora el estado general, desaparecen los dolores y la inflamación articular. La febrícula había desaparecido antes de comenzar el tratamiento con corticosteroides. La uveítis desapareció rápidamente.
Se recibió el informe final de anatomía patológica de un ganglio mediastinal con el diagnóstico definitivo de sarcoidosis
Sarcoidosis
Introducción:
La sarcoidosis es un trastorno granulomatoso sistémico de etiología desconocida, caracterizado por la presencia de granulomas no caseosos en los órganos afectados. Típicamente afecta adultos jóvenes, e inicialmente se presenta con una o más de las siguientes anormalidades:
Introducción:
La sarcoidosis es un trastorno granulomatoso sistémico de etiología desconocida, caracterizado por la presencia de granulomas no caseosos en los órganos afectados. Típicamente afecta adultos jóvenes, e inicialmente se presenta con una o más de las siguientes anormalidades:
* Adenopatías hiliares bilaterales.
* Opacidades reticulares pulmonares.
* Lesiones de piel, articulaciones u ojos.
Epidemiología:
La prevalencia es de aproximadamente es de 10 a 20 por 100.000, y la incidencia anual no se conoce.
Los negros tienen más predisposición a padecerla. Además, hay un "background" inmunogénico, que puede jugar un rol en las manifestaciones clínicas de la sarcoidosis. Por ejemplo, los negros suelen tener formas más agresivas que los blancos.
Hay numerosos reportes de familias con muchos miembros que padecen la enfermedad. El hallazgo más prominente de vínculo se ve a nivel del antígeno mayor de histocompatibilidad (MHC o HLA) en el brazo corto del cromosoma 6 Parece ser que varios alelos confieren susceptibilidad a padecer la enfermedad (por ej HLA DR 11, 12,14,15,17) y otros parecen protectores (ej HLA DR1, DR4, y posiblemente DQ*0202)
Etiología:
La etiología y patogenia son desconocidas. Sin embargo, existen varias hipótesis sobre la misma:
A pesar de los avances en el conocimiento de la inmunopatogénesis de la sarcoidosis, el estímulo antigénico que inicia la enfermedad permanece desconocido. Sin embargo, se han inculpado a algunos tipos de exposición ocupacional, dentro de las cuales las sales de berilio han mostrado que producen granulomas similares a los vistos en la sarcoidosis.
Una serie de casos sugiere que la exposición al polvo del World Trade Center después del desastre del 11 S, produjo entre las personas que colaboraron en el rescate o aquellos rescatados, un aumento de la incidencia de sarcoidosis durante los 5 años posteriores al desastre.
Dentro de los agentes infecciosos que han sido inculpados, están principalmente las micobacterias sobre todo a Mycobacterium tuberculosis. El Propionibacterium acnes, y el herpes virus humano tipo-8
Patología:
La lesión inicial dentro del sistema pulmonar es una alveolitis por células T CD4(+), seguido por la formación de un granuloma no caseoso. Los granulomas tienen un centro cerrado compuesto de macrófagos, células epitelioides, y células gigantes multinucleadas, rodeadas por linfocitos, monocitos, células cebadas, y fibroblastos. Dentro del pulmón, los granulomas tienden a ubicarse de una manera broncocéntrica, lo que puede conducir a obstrucción o restricción pulmonar.
El granuloma sarcoide puede resolverse sin secuela, o puede sufrir una fibrosis obliterativa, con el resultado de una fibrosis intersticial
Manifestaciones Clínicas:
La sarcoidosis se presenta en el 70 a 90% de los casos, en pacientes de entre 10 y 40 años de edad. En la mitad de los casos la enfermedad se detecta incidentalmente por alteraciones radiológicas (por ej adenopatías hiliares bilaterales, opacidades intersticiales pulmonares) en una Rx de tórax de rutina, antes de presentar síntomas.
La sarcoidosis afecta principalmente pulmón, y los síntomas comunes de presentación son tos, disnea y dolor torácico. Las manifestaciones extrapulmonares son fundamentalmente a nivel de piel y ojos.
Otros síntomas incluyen fatiga, malestar, fiebre y pérdida de peso.
En el examen físico no es común auscultar rales, pero si sibilancias.
Imágenes Pulmonares:
El compromiso pulmonar ocurre en 90% de los pacientes. El patrón clásico revela adenopatías hiliares bilaterales. Este hallazgo, sin embargo, puede estar ausente, o si está presente, puede ocurrir en combinación con opacidades parenquimatosas. Las opacidades parenquimatosas pueden ser intersticiales, alveolares, o ambas. El compromiso de la pleura es raro (menos del 5% de los casos), pero puede, cuando se presenta ser un derrame pleural exudativo linfocitario, quilotórax, hemotórax o neumotórax.
Se han descripto 4 estadios en el compromiso pulmonar de la sarcoidosis.
Estadio I.
Es definido por la presencia de adenopatías hiliares bilaterales, que a menudo se acompaña de agrandamiento ganglionar paratraqueal derecho. El 50% de los pacientes muestran adenopatías hiliares bilaterales como primera expresión de sarcoidosis. La regresión de los ganglios hiliares dentro de 1 a 3 años, ocurre en 75% de los pacientes. Hay un 10% que persisten con adenopatías hiliares crónicas por 10 años o más.
Estadio II.
Consiste en adenopatías hiliares bilaterales y opacidades reticulares (estas últimas se observan en las bases pulmonares). Solo el 25% de los pacientes debutan en este estadio. Usualmente los pacientes tienen tos, disnea, fiebre, y fatiga fácil.
Estadio III.
Consiste en opacidades reticulares parenquimatosas con desaparición o achicamiento de las adenopatías hiliares. Las opacidades reticulares se observan predominantemente en las zonas superiores.
Estadio IV.
Se caracteriza por opacidades reticulares, con evidencia de pérdida de volumen, predominantemente distribuidas en las zonas superiores de ambos pulmones. Puede haber conglomerados en forma de masas con marcadas bronquiectasias por tracción. También puede verse cavitación o formación de quistes.
Sarcoidosis nodular.
En esta forma de presentación radiológica la Rx muestra múltiples nódulos bilaterales, y mínimas adenopatías hiliares, hallazgo que suele confundir con metástasis pulmonares. En la TAC se ven nódulos de bordes mal definidos.
Tomografía Computada.
La TAC muestra una variedad de alteraciones en pacientes con sarcoidosis:
Tomografía Computada.
La TAC muestra una variedad de alteraciones en pacientes con sarcoidosis:
* Linfadenopatía hiliar bilateral.
* Arrosariamiento o engrosamiento irregular de la trama broncovascular.
* Nódulos siguiendo bronquios, vasos, y regiones subpleurales.
* Engrosamiento de las paredes bronquiales.
* Opacidades pulmonares en “vidrio esmerilado”.
* Consolidación o masas parenquimatosas.
* Trazos fibrosos parenquimatosos.
* Quistes.
* Bronquiectasias por tracción.
* Fibrosis con distorsión de la arquitectura pulmonar.
La TAC de alta resolución pulmonar revela que las lesiones afectan más comúnmente las zonas medias y superiores de los pulmones. Estas alteraciones pueden no ser vistas en la Rx de tórax.
Los hallazgos en la TAC de alta resolución pueden correlacionarse con los hallazgos histológicos. Por ejemplo las opacidades en “vidrio esmerilado” se asocian a granulomas sarcoides más que a alveolitis.
* Arrosariamiento o engrosamiento irregular de la trama broncovascular.
* Nódulos siguiendo bronquios, vasos, y regiones subpleurales.
* Engrosamiento de las paredes bronquiales.
* Opacidades pulmonares en “vidrio esmerilado”.
* Consolidación o masas parenquimatosas.
* Trazos fibrosos parenquimatosos.
* Quistes.
* Bronquiectasias por tracción.
* Fibrosis con distorsión de la arquitectura pulmonar.
La TAC de alta resolución pulmonar revela que las lesiones afectan más comúnmente las zonas medias y superiores de los pulmones. Estas alteraciones pueden no ser vistas en la Rx de tórax.
Los hallazgos en la TAC de alta resolución pueden correlacionarse con los hallazgos histológicos. Por ejemplo las opacidades en “vidrio esmerilado” se asocian a granulomas sarcoides más que a alveolitis.
PET scan.
El PET scan con 18-fluorodeoxiglucosa, puede ayudar a identificar lesiones ocultas y posiblemente enfermedad granulomatosa reversible. Este método no discrimina entre neoplasia y sarcoidosis ya que puede ser positivo en ambos procesos.
Scan con Galio-67
Scan con Galio-67
es un test no invasivo, útil para estadificar la alveolitis.
Sarcoidosis Extrapulmonar.
Piel:
El compromiso cutáneo se ve en 20% de los pacientes con sarcoidosis y a menudo es un hallazgo temprano.
Puede haber rash máculo-papular que es la lesión subaguda más común. Usualmente compromete alas de la nariz, labios, párpados, frente, región de cuello, y sitios con trauma previo (cicatrices o tatuajes).
Frecuentemente hay lesiones como placas de color violáceo, llamadas lupus pernio, sobre todo en sarcoidosis crónica, en nariz, mejillas, mentón y orejas.
Eritema nodoso es una paniculitis, que puede formar parte del síndrome de Lofgren de muy buen pronóstico.
Puede haber lesiones atípicas ulcerativas, psoriasiformes, hipopigmentadas, foliculares, angiolupoides, rosácea-like, o de tipo morfea.
Lesiones Oftalmológicas.
Un 20% de los pacientes presentan lesiones oculares. Estas lesiones incluyen:
Uveítis anterior (iridociclitis), uveítis posterior (coriorretinitis), vasculitis retiniana, queratoconjuntivitis, folículos conjuntivales.Además, el glaucoma secundario, cataratas, y ceguera son complicaciones tardías. La combinación de uveítis anterior, agrandamiento parotídeo, parálisis facial y fiebre (fiebre uveoparotídea) se conoce como síndrome de Heerfordt.Los tejidos extraoculares pueden afectarse como las glándulas lagrimales, los músculos extraoculares, y la vaina del nervio óptico, además de poder presentarse como una masa retroorbitaria de partes blandas.
Sistema Reticuloendotelial.
Puede manifestarse como:
* Linfadenopatía periférica (hasta el 40% de los pacientes).
* Hepatomegalia (hasta el 20%).
* Granulomas no caseosos en la biopsia hepática con o sin hepatomegalia (75%).
* Esplenomegalia (25 a 80% de los casos de los que tienen granulomas).
* Hiperesplenismo, que puede llevar a anemia, trombocitopenia y leucopenia.
Musculoesquelético.
El sistema musculoesquelético puede ocurrir en hasta el 10% de los pacientes con sarcoidosis y pueden incluir:
Poliartritis aguda (especialmente tobillos), usualmente asociado a eritema nodoso y ocasionalmente asociado a uveítis aguda.Poliartritis crónica con reabsorción ósea perióstica. Radiográficamente parecen quistes, que pueden generar confusion con artritis reumatoidea. No hay correlación entre lesiones óseas y calcemia.
Miositis granulomatosa difusa, es una complicación rara de la sarcoidosis.
Síndrome de Lofgren.
Es la combinación de eritema nodoso, adenopatías hiliares, poliartralgias migratorias, y fiebre, visto generalmente en mujeres jóvenes. En general, el síndrome de Lofgren está asociado a buen pronóstico y a remisión espontánea.
Glándulas exócrinas.
Inflamación indolora de glándulas salivales ocurre en 4% de los pacientes con sarcoidosis. Se puede ver también xerostomía y queratoconjuntivitis sicca, similares a los vistos en el síndrome de Sjögren. Puede haber pancreatitis.
Riñón y electrolitos.
Las alteraciones en el metabolismo del calcio son las más comunes. El defecto en el metabolismo del calcio es debido a la producción extrarrenal de calcitriol por los macrófagos activados.Hay aumento de la absorción intestinal de calcio, hipercalciuria (que ocurre en hasta 50% de los casos), hipercalcemia(que ocurre en 10 a 20% de los casos), y nefrocalcinosis. Si no es tratada la deposición de calcio renal puede conducir a insuficiencia renal crónica.
Aunque la infiltración parenquimatosa del riñón no es infrecuente, raramente es causa de disfunción renal por si sola. Otras complicaciones renales de la sarcoidosis incluyen la nefropatía membranosa, una glomerulonefritis con formación de semilunas, glomerulosclerosis, poliuria (debido a diabetes insípida central o nefrogénica), hipertensión y una variedad de defectos tubulares.
Cardiovascular.
El compromiso granulomatoso del septum ventricular y del sistema de conducción puede conducir a arritmias, incluyendo bloqueo cardíaco completo y muerte súbita. Tal compromiso puede ser precedido por palpitaciones, síncope, mareos, o dolor torácico.Además, la hipertensión pulmonar puede sobrevenir, siendo consecuencia de severa cicatrización del parénquima pulmonar y obliteración pulmonar. En este contexto, la muerte por sarcoidosis usualmente resulta de fallo ventricular derecho.
Neurológico.
Aproximadamente 5% de los pacientes con sarcoidosis tienen compromiso neurológico, que puede, ocasionalmente ser el síntoma de presentación.
Las manifestaciones del sistema nervioso central ocurren tempranamente, mientras que los nervios periféricos se ve característicamente en estadios tardíos.
La meningitis granulomatosa de la base, con infiltración o compresión de estructuras adyacentes, es responsable de la mayoría de las manifestaciones del compromiso del sistema nervioso central incluyendo:
* Hipopituitarismo hipotalámico.
* Diabetes insípida central.
* Hidrocefalia.
* Meningitis linfocitaria. Parálisis de nervios craneales, particularmente parálisis facial.
Gastrointestinal.
Ocurre en 0,1 a 0,9% de los pacientes con sarcoidosis, aunque la incidencia de compromiso subclínico es mucho mayor. El estómago es la porción del tracto gastrointestinal más comprometido, pero se han descripto también la sarcoidosis de esófago, apéndice, colon, y recto. Puede también afectar el hígado y páncreas. El compromiso del intestino delgado ha sido reportada pero es muy rara.
Sistema reproductivo.
Hay reportes de compromiso de endometrio, ovario, o útero detectado durante la evaluación y tratamiento de metrorragias o masas pélvicas. La sarcoidosis generalmente no afecta la fertilidad, excepto cuando hay compromiso cardiopulmonar severo. Suele mejorar durante el embarazo, probablemente por aumento de los niveles de cortisol libre materno.
Puede comprometer testículos, y debe ser diferenciado del cáncer testicular y de la tuberculosis. La epididimitis recurrente raramente ocurre.
Tiroides.
Puede causar bocio difuso, o raramente un nódulo solitario. Casi todos los pacientes son eutiroideos, aunque algunos casos de hipotiroidismo por reemplazo del tejido tiroideo han sido reportados.
Fatiga.
Es un síntoma común en pacientes con sarcoidosis. Un estudio encontró mayor gasto energético en reposo y un nivel elevado de proteína C reactiva (PCR) en un subset de pacientes con fatiga.
Alteraciones de laboratorio
Una variedad de alteraciones de laboratorio están presentes en la sarcoidosis:
Anemia no es infrecuente, generalmente de los procesos crónicos aunque puede haber hiperesplenismo, o anemia hemolítica autoinmune.
Leucopenia (5 a 10%), eosinofilia (25%) y trombocitopenia (rara) pueden ser vistos.
La eritrosedimentación está frecuentemente elevada, pero no es útil en el seguimiento de la actividad de la enfermedad.
Hipercalciuria es más comúnmente observada en la hipercalcemia.
Hipergamaglobulinemia (30 a 80%), reactividad de los tests cutáneos disminuida, y test de factor reumatoideo positivo pueden existir.Una moderada elevación de la fosfatasa alcalina generalmente es indicativo de compromiso granulomatoso hepático.
Los gases en sangre pueden ser normales, o raramente haber hipoxemia e hipocapnia (hiperventilación) El test de ejercicio puede acentuar esas anormalidades.
Nivel sérico de enzima convertidora de angiotensina (ECA) está elevada en 75% de los pacientes con sarcoidosis no tratada. Los resultados falsos positivos son inusuales (menos de 10%), pero lo suficientemente frecuentes para limitar la utilidad de la ECA como test diagnóstico. El valor de ECA tisular es típicamente muy alto. La utilidad en el seguimiento de la actividad de la enfermedad no está claro.
Tests de función pulmonar.
Característicamente revelan un patrón restrictivo con reducción de la capacidad de difusión de CO, aunque no es inusual que los tests de función pulmonares sean normales. La sarcoidosis con compromiso endobronquial puede dar un patrón obstructivo. El mayor valor de estos tests es medir el curso de la enfermedad en pacientes individuales por medidas secuenciales.
Lavado broncoalveolar (BAL)
Puede usarse como medida agregada a la batería de estudios diagnósticos de sarcoidosis, por demostrar un número reducido de células CD8 y elevación de CD4, con aumento de la relación CD4/CD8, con un aumento de las células T activadas, inmunoglobulinas, y células secretoras de IgG. Puede ayudar en descartar infección como diagnóstico alternativo. El hallazgo de linfocitosis en el BAL no es específico ni sensible para el diagnóstico de sarcoidosis. Hay hallazgos en el BAL que tienen interés, por ejemplo: en un líquido de BAL con más de 2% de neutrófilos o más de 1% de eosinófilos descarta sarcoidosis. Una relación CD4/CD8 menor de 1, tiene un valor predictivo negativo de 100% para sarcoidosis. Sin embargo, estos resultados deben ser interpretados con precaución, sobre todo con recientes reportes de sarcoidosis con predominancia de CD8. La tríada de relación de CD4 a CD8 mayor de 4, un porcentaje de linfocitos mayor o igual a 16%, y una biopsia transbronquial que muestra granulomas no-caseosos fueron los tests más específicos para sarcoidosis. Esta combinación de hallazgos se asoció con 100% de valor predictivo positivo para distinguir sarcoidosis de otras enfermedades intersticiales pulmonares, y 81% de valor predictivo positivo para distinguir sarcoidosis de otras enfermedades.
El dímero-D en el BAL también sostiene el diagnóstico de sarcoidosis. Un estudio observacional encontró que 8 de cada 10 pacientes con sarcoidosis tienen dímero-D detectable en el líquido de BAL (más de 78 ng de dímero-D/ml de líquido concentrado de BAL) comparado con ninguno de 18 pacientes control.
Histopatología.
La característica morfológica típica de la sarcoidosis es el granuloma no caseoso en pulmón, que es más comúnmente encontrado en los septos alveolares, las paredes de los bronquios, y las arterias y venas pulmonares.
La formación de granulomas es probablemente precedida por una alveolitis que afecta el intersticio más los espacios alveolares y que se caracteriza por la acumulación de células inflamatorias, incluyendo monocitos, macrófagos, y linfocitos.
El granuloma es una reacción inflamatoria crónica focal formada por acumulación de células epiteliales, monocitos, linfocitos, macrófagos y fibroblastos. Las células gigantes multinucleadas son frrecuentemente encontradas entre las células epitelioides dentro de los folículos del folículo del granuloma y a menudo tienen inclusiones citoplasmáticas, tales como los cuerpos asteroides, los cuerpos de Schaumann, y las partículas cristalinas birrefringentes (oxalato de calcio y otras sales de calcio) La mayoría de los granulomas sarcoides se resuelven y dejan poca o ninguna manifestación residual de inflamación previa.
Diagnóstico
No existe un test diagnóstico definitivo para sarcoidosis. En cambio, el diagnóstico de sarcoidosis requiere 3 elementos:
El dímero-D en el BAL también sostiene el diagnóstico de sarcoidosis. Un estudio observacional encontró que 8 de cada 10 pacientes con sarcoidosis tienen dímero-D detectable en el líquido de BAL (más de 78 ng de dímero-D/ml de líquido concentrado de BAL) comparado con ninguno de 18 pacientes control.
Histopatología.
La característica morfológica típica de la sarcoidosis es el granuloma no caseoso en pulmón, que es más comúnmente encontrado en los septos alveolares, las paredes de los bronquios, y las arterias y venas pulmonares.
La formación de granulomas es probablemente precedida por una alveolitis que afecta el intersticio más los espacios alveolares y que se caracteriza por la acumulación de células inflamatorias, incluyendo monocitos, macrófagos, y linfocitos.
El granuloma es una reacción inflamatoria crónica focal formada por acumulación de células epiteliales, monocitos, linfocitos, macrófagos y fibroblastos. Las células gigantes multinucleadas son frrecuentemente encontradas entre las células epitelioides dentro de los folículos del folículo del granuloma y a menudo tienen inclusiones citoplasmáticas, tales como los cuerpos asteroides, los cuerpos de Schaumann, y las partículas cristalinas birrefringentes (oxalato de calcio y otras sales de calcio) La mayoría de los granulomas sarcoides se resuelven y dejan poca o ninguna manifestación residual de inflamación previa.
Diagnóstico
No existe un test diagnóstico definitivo para sarcoidosis. En cambio, el diagnóstico de sarcoidosis requiere 3 elementos:
* Manifestaciones clínico-radiográficas compatible.
* Exclusión de otras enfermedades que se puedan presentar de manera similar.
* Detección histopatológica de granulomas no caseosos.
Estos elementos se logran por una evaluación exhaustiva de todo paciente con sarcoidosis sospechada, seguida de procedimientos diagnósticos en la mayoría de los casos.
Las enfermedades que también pueden presentarse con alteraciones radiológicas predominantemente en lóbulo superior de características alveolares, intersticiales, nodulares, o quísticas, incluyen: neumonitis por hipersensibilidad, granuloma eosinofílico, enfermedad colágeno vascular, neumoconiosis, enfermedad pulmonar crónica por berilio (beriliosis), e infecciones, particularmente tuberculosis e histoplasmosis.
Afortunadamente, la mayoría de los casos son fácilmente reconocidos, y crean mínimos dilemas diagnósticos. Hay, sin embargo, situaciones donde es difícil la interpretación y el diagnóstico puede no ser tan claro. Por ejemplo, el diagnóstico puede ser dificultoso en ausencia de manifestaciones extrapulmonares, marcada linfadenopatía hiliar, o mediastinal. En forma similar, la presencia de infección por HIV, una exposición ocupacional al berilio, o agentes infecciosos, y/o prominentes síntomas sistémicos (por ejemplo fiebre o sudores nocturnos, y/o fatiga) aumenta la preocupación por infección o neoplasia.
Los pacientes con inmunodeficiencia común variable, pueden tener un cuadro sarcoidosis-like, con infiltración granulomatosa del hígado y pulmones, y elevaciones variables de ECA. Por lo tanto, los pacientes con aparente sarcoidosis, que tienen hipogamaglobulinemia y tienen infecciones recurrentes deben ser evaluados para descartar dicha entidad.
Evaluación inicial.
En todo paciente en quien se sospeche sarcoidosis se debe hacer una amplia evaluación inicial. El propósito de la misma busca obtener datos adicionales que confirmen el diagnóstico, y descarten otras alternativas.
Dicha evaluación, la mayoría de las veces comprende lo siguiente:
* Historia Clínica exhaustiva, incluyendo exposición ambiental y ocupacional.
* Examen Físico Completo.
* Rx de tórax.
* Tests de función pulmonar, incluyendo espirometría, y difusión de monóxido de carbono (DLCO).
* Recuentos citológicos en sangre periférica.
* Exámenes de rutina de laboratorio.
* Análisis de orina.
* Electrocardiograma.
* Examen oftalmológico.
* Test cutáneo de tuberculina.
Otros tests pueden ser considerados como nivel de enzima convertidora de angiotensina (ECA), y lavado broncoalveolar (BAL).
Procedimientos diagnósticos.
Los pacientes que se presentan con el clásico síndrome de Lofgren, fiebre, eritema nodoso, y linfadenopatía hiliar bilateral, pueden no requerir biopsia, sobre todo si las manifestaciones se resuelven rápida y espontáneamente, y no hay explicación alternativa para el cuadro. Para todos los demás pacientes con sospecha de sarcoidosis, la biopsia para confirmar el diagnóstico está indicada.
Las biopsias deben ser llevadas a cabo en lesiones accesibles, que pueden ser ganglios palpables, nódulos subcutáneos, lesiones cutáneas, glándulas parótidas o lagrimales agrandadas. El eritema nodoso no debe ser biopsiado debido a que la histopatología demostrará paniculitis y no granulomas, aún cuando la causa del mismo sea sarcoidosis.
La fibrobroncoscopía con biopsia de pulmón transbronquial (o biopsia de lesiones endobronquiales visibles) es actualmente el procedimiento preferido si no hay lesiones periféricas fácilmente accesibles. Las muestras deben enviarse tanto para cultivos como para examen histopatológico, incluyendo tinciones para hongos y bacilos ácido alcohol resistentes.
La aspiración transbronquial con aguja guiada por ultrasonografía puede ayudar a mejorar el rédito diagnóstico de una aspiración con aguja fina de un ganglio mediastinal. Esas técnicas pueden evitar una biopsia quirúrgica en algunos pacientes, pero requieren un equipamiento especial y una experiencia significativa del operador.
La biopsia de pulmón a cielo abierto, o biopsia pulmonar toracoscópica, o una biopsia ganglionar mediastinal deben llevarse a cabo si otros tests menos invasivos no proveen evidencias diagnósticas.
Monitoreo.
No hay datos sobre las indicaciones de tests específicos ni de la óptima frecuencia de monitoreo de la actividad de la enfermedad. Los pacientes con síntomas más significativos al comienzo necesitarán evaluación más frecuente, y aquellos con síntomas mínimos o ningún síntoma las evaluaciones deben ser más espaciadas. Por ejemplo, pacientes que comienzan con prednisona para enfermedad activa necesitarán ser reevaluados en intervalos de 4 a 8 semanas., pero los pacientes asintomáticos pueden ser vistos con intervalos de 3 a 4 meses por el primer año, y menos frecuentemente de allí en adelante.
Tratamiento de la Sarcoidosis Pulmonar.
El tratamiento de la sarcoidosis pulmonar permanece controversial por las siguientes razones:
Un gran número de pacientes tienen remisiones espontáneas o tienen un curso clínico benigno. Se ha estimado que estas remisiones ocurren en aproximadamente 60 a 80% de los pacientes en estadio I, 50 a 60% en estadio II, y menos de 30% en estadio III.
No hay forma fácil de evaluar actividad y severidad de la enfermedad, por lo que el curso y el pronóstico de la misma es dificultoso.
La marcada variabilidad en la presentación y curso clínico hace dificultoso desarrollar guías de tratamiento. La causa de la enfermedad es desconocida, y por lo tanto no existe tratamiento específico.
Pocos estudios han sido llevados a cabo para contestar muchas de las siguientes preguntas en referencia al manejo de la sarcoidosis pulmonar, tales como:
Cuales son las indicaciones para el tratamiento?
Cuándo debe ser comenzada la terapia?
Cuál es la mejor terapia?
Es beneficioso el tratamiento a largo plazo?
Cómo debe ser monitoreado el curso de la enfermedad?
El tratamiento, altera el curso del proceso granulomatoso?
La actual terapia de la sarcoidosis apunta a reducir la respuesta inflamatoria, reduciendo la “carga de granulomas” y previniendo el desarrollo de fibrosis.
Dado la capacidad de atenuar la respuesta inflamatoria, los glucocorticoides son considerados capaces de detener o disminuir la progresión de la fibrosis pulmonar que puede desarrollar en la sarcoidosis. Como resultado, los glucocorticoides han sido los agentes más comúnmente usados para el tratamiento de la sarcoidosis pulmonar. Basados en la evidencia actual, su uso en este contexto está justificado para alivio de los síntomas y para controlar el compromiso sistémico.
Mecanismo de acción de los corticosteroides.
Los fagocitos mononucleares y los linfocitos T juegan un rol clave en la patogénesis de la sarcoidosis, y los glucocorticoides alteran la acción de ambas células.
Los glucocorticoides entran al citoplasma y se unen a sus receptores (formas inactivas unidas a moléculas de proteínas de choque térmico 90 kD) (HSP, del inglés Heat Shock Proteins). Siguiendo a la unión, las moléculas de las proteínas del choque térmico se disocian, y de esa manera exponen un dominio de unión a DNA. El complejo esteroide-receptor entra al núcleo y se une a los elementos de respuesta glucocorticoidea (localizados “corriente arriba” de la región promotora del gen), lo que resulta en la modulación de la transcripción génica.
Este efecto en la transcripción génica induce la síntesis de IkBa, una proteína que atrapa y de ese modo inactiva al factor nuclear kappa B (NF-κB). Esta última proteína es un activador de genes de citoquinas y un mediador de la acción proinflamatoria del factor de necrosis tumoral (TNF). La inactivación de este factor nuclear resulta en la inhibición de la síntesis de casi todas las citoquinas conocidas, produciéndose así el mayor efecto inmunosupresor de los corticosteroides. Entre las citoquinas inhibidas que son consideradas patogénicamente importantes en la sarcoidosis están el factor de necrosis tumoral-alfa (TNF-alfa), interleukina -1 (IL-1), factor estimulante de colonias granulocíticas, interleukina-2 (IL-2), e interferon-gama (IF- gama). La síntesis de prostaglandina E2 está también disminuida.
Uso racional de los corticosteroides y selección de pacientes.
El tratamiento de la sarcoidosis debe basarse en evidencias clínicas y de laboratorio de disfunción, que puede estar relacionada con manifestaciones pulmonares y extrapulmonares de sarcoidosis.
Indicaciones para el tratamiento de la sarcoidosis pulmonar.
Las indicaciones usuales para la terapia de la sarcoidosis pulmonar son:
* Empeoramiento de los síntomas pulmonares incluyendo. Tos, dificultad respiratoria, dolor torácico, y hemoptisis.
* Deterioro de la función pulmonar cuando es medida con tests seriados a intervales de 3 a 6 meses que demuestran uno o más de los siguientes: una caída en la capacidad pulmonar total de 10% o más; una caída en la capacidad vital forzada de 15% o más; una disminución de la capacidad de difusión de 20% o más; o empeoramiento del intercambio de gases en reposo o en ejercicio.
* Cambios radiográficos progresivos, incluyendo: empeoramiento de las opacidades intersticiales, desarrollo de cavidades, progresión de la fibrosis con panalización, o desarrollo de hipertensión pulmonar.
La terapia no está indicada en los siguientes grupos de pacientes:
* Pacientes asintomáticos en estadio I (linfadenopatía hiliar con o sin eritema nodoso). Tales pacientes tienen una alta tasa de remisiones espontáneas.
* Pacientes asintomáticos en estadio II con función pulmonar normal o levemente anormal (leve restricción u obstrucción con intercambio de gase normales). Esos pacientes deben ser seguidos por 3 a 6 meses para demostrar algún grado de progresión de la enfermedad antes de comenzar tratamiento. Es importante en estos pacientes, documentar un deterioro de la función pulmonar o del intercambio de gases en este contexto antes de comenzar el tratamiento, debido a que aproximadamente 50% de los pacientes con estadio II radiográfico se resuelven espontaneamente en 36 meses.
* Los pacientes asintomáticos en estadio III y función pulmonar normal o casi normal deben ser seguidos de cerca por 3 a 6 meses. Sin embargo, debido a que solo el 33% de estos pacientes con estadio III radiográfico muestran resolución después de 5 años, la mayoría requerirá terapia.
Indicaciones para Tratamiento de Sarcoidosis Extrapulmonar.
Otras indicaciones para la terapia incluyen severo malestar o incapacidad para trabajar como resultado de fiebre, fatiga, artralgia, neuropatía enfermedad cutánea desfigurante, enfermedad de vías aéreas superiores, o insuficiencia hepática. El tratamiento de la sarcoidosis ocular, neurológica, miocárdica o renal, o la hipercalcemia está indicada aún cuando los síntomas son leves, debido a pérdida severa de la visión, arritmias fatales, o daño renal insidioso.
Protocolo de Uso de los Corticosteroides.
La dosis óptima de corticosteroides no se conoce, así que la dosis requiere balancear los riesgos de efectos adversos con la probabilidad de respuesta. Teóricamente uno desea elegir la menor dosis necesaria para obtener óptimos beneficios.
La terapia se inicia con relativamente alta dosis de corticosteroides, seguido por una reducción lenta hasta la menor dosis efectiva con una duración de entre 6 y 12 meses.
Las primeras 4 a 6 semanas se comienza con una dosis de 0,5 a 1 mg/kg de peso ideal (usualmente 30 a 60 mg/día).
Después de 4 a 6 semanas, el paciente debe ser reevaluado. Si la condición es estable o mejorada, la dosis se baja alrededor de 5 a 10 mg cada 4 a 8 semanas hasta llegar a 15 a 30 mg/día.
Si el paciente continua estable o mejorado, la dosis continúa bajándose hasta que una dosis de mantenimiento es alcanzada (aproximadamente 0,25 mg/kg de peso ideal por día, usualmente 10 a 15 mg/día.
Dado que las recidivas de síntomas tales como tos, disnea y dolor torácico son comunes (ocurren en 60% de los pacientes) se recomienda que la dosis de mantenimiento se continúe hasta 6 a 8 meses, con una duración total del tratamiento de alrededor de 1 año. Frecuentemente un breve curso de mayores dosis (aumentos de 10 a 20 mg por encima de la dosis de mantenimiento dadas por 2 a 4 semanas) son requeridas para aliviar la recurrencia de los síntomas.
Hay esquemas de días alternos que no han mostrado beneficios respecto de las dosis diarias.
Altas Dosis de Corticosteroides Orales.
En pacientes con compromiso cardíaco, neurológico, ocular o de vías aéreas superiores dosis de 80 a 100 mg/día pueden ser requeridas.
Los corticosteroides inhalados se pueden usar en el tratamiento de la sarcoidosis
pulmonar para modular la alveolitis y proveen beneficios clínicos en algunos pacientes. Se ha usado fluticasona 2000 ug/día, budesonida 800 a 1600 mcg dos veces por día etc.
Midiendo la Respuesta a la Terapia.
Para comprender el efecto de la terapia en el curso de la sarcoidosis, es importante reconocer aquellos factores que influencian el pronóstico de sarcoidosis, independientemente de la terapia. Los elementos de pronóstico favorable incluyen:
* Enfermedad en estadio I especialmente cuando está asociado a eritema nodoso.
* Ausencia de síntomas.
En contraste, los elementos de mal mall pronóstico incluyen:
* Presencia de síntomas y compromiso multisistémico (tres o más órganos comprometidos).
* Disnea, es aveces indicativa de disfunción pulmonar irreversible.
* Lesiones asociadas de piel, óseas (quistes), o artritis (especialmente si la edad es más de 30 años).
*Descendencia Africana.
* Aumento de los infiltrados pulmonares.
Aún teniendo en cuenta todos estos factores pronósticos, es a menudo dificultoso determinar si el tratamiento está produciendo el efecto deseado. Una respuesta favorable a los corticosteroides es definida por:
* Una disminución de los síntomas, especialmente disnea, tos, hemoptisis, dolor torácico, o fatiga.
* Una reducción de las manifestaciones radiográficas.
* Un mejoramiento fisiológico, incluyendo 10 a 15% de mayor aumento en la FVC o TLC, un 20% de aumento en la DLCO, o un mejoramiento en el intercambio gaseoso (un aumento mayor de 4 mm Hg en la PO2 arterial).
Fuente: UpToDate
Autores
Talmadge E King, Jr, MD
Andrew Fontenot, MD
Kevin R Flaherty, MD, MS
Andrew Nicholson, MD
Helen Hollingsworth, MD