Mujer de 73 años con síndrome coronario y bicitopenia. ¡Lo invitamos a opinar!
Motivo de consulta: Dolor torácico
Enfermedad actual:
Mujer de 76 años de edad, consulta por dolor retroesternal, de tipo opresivo, de 5 horas de evolución, de inicio súbito, intensidad 8/10, sin irradiación, que cede con nitritos, acompañado de disnea de reposo y episodio de pérdida deconocimiento sin relajación de esfínteres. Por este cuadro ingresa a Unidad Coronaria (UCO) donde se constata un síndrome coronario agudo (angina inestable).
Durante la internación en UCO se constata fiebre y tos con expectoración mucopurulenta, asociadas a bicitopenia (neutropenia y anemia) e insuficiencia renal. Por este cuadro inicia tratamiento con ampicilina sulbactam de forma empírica. Resuelto el síndrome coronario agudo, pasa a sala general a cargo del Servicio de Clínica Médica.
Durante la internación en UCO se constata fiebre y tos con expectoración mucopurulenta, asociadas a bicitopenia (neutropenia y anemia) e insuficiencia renal. Por este cuadro inicia tratamiento con ampicilina sulbactam de forma empírica. Resuelto el síndrome coronario agudo, pasa a sala general a cargo del Servicio de Clínica Médica.
Antecedentes:
1 Hipertensión arterial, diagnosticada hace 36 años, en tratamiento desde hace 6 años con atenolol 50 mg/día, losartan 50 mg/día, clortalidona 50 mg/día y furosemida 40 mg/día
2 Cardiopatía isquémica (IAM) hace 6 años, en tratamiento con mononitrato de isosorbide 20 mg/día, ácido acetil salicílico 100 mg/día y clopidogrel 75 mg/día
3 Dislipidemia, desde hace 3 años, en tratamiento con atorvastatina 10 mg/día
4 Histerectomía por leiomioma uterino hace 30 años
5 Artralgias desde hace 20 años, a nivel de hombros, codos, muñecas, manos, rodillas y tobillos, asociadas a rigidez matinal, que cedían en forma parcial con la administración de AINES
1 Hipertensión arterial, diagnosticada hace 36 años, en tratamiento desde hace 6 años con atenolol 50 mg/día, losartan 50 mg/día, clortalidona 50 mg/día y furosemida 40 mg/día
2 Cardiopatía isquémica (IAM) hace 6 años, en tratamiento con mononitrato de isosorbide 20 mg/día, ácido acetil salicílico 100 mg/día y clopidogrel 75 mg/día
3 Dislipidemia, desde hace 3 años, en tratamiento con atorvastatina 10 mg/día
4 Histerectomía por leiomioma uterino hace 30 años
5 Artralgias desde hace 20 años, a nivel de hombros, codos, muñecas, manos, rodillas y tobillos, asociadas a rigidez matinal, que cedían en forma parcial con la administración de AINES
Antecedentes familiares:
1 Padre fallecido de cáncer de pulmón.
2 Esposo fallecido de tuberculosis pulmonar hace 15 años.
Hábitos:
1 Alcohol: niega
2 Tabaco: refiere 40 a 50 cigarrillos por día durante 30 años, con abandono hace 13 años
3 Otra medicación: cilostazol 100 mg/día, ranitidina 150 mg/12 hs, carbonato de calcio y diclofenac 50 mg/8 horas
Examen físico:
1 Vigil y orientada globalmente
2 Signos vitales: PA 130/70 mmHg; FC 75 lpm; FR 12 rpm; Temperatura 36,5 º C
3 Cabeza y cuello: conjuntivas pálidas, escleras blancas. No se palpan adenopatías ni tiroides.
4 Tórax: mamas sin tumoraciones ni lesiones evidentes.
5 Aparato respiratorio: expansión y excursión conservadas. Murmullo vesicular disminuido en forma generalizada con rales crepitantes en base izquierda.
6 Aparato cardiovascular: ruidos normofonéticos, soplo sistólico 3/6 en foco aórtico con irradiación al mesocardio y cuello. Pulsos periféricos conservados.
7 Abdomen: blando, depresible e indoloro. Hígado palpable 2 cm por debajo del reborde costal.
8 Miembros: desviación y deformidad a nivel de las falanges distales. Dolor a la movilización activa y pasiva de ambos hombros, codos, muñecas, dedos de las manos, rodillas y tobillos. No se palpan adenopatías ni edema.
9 Neurológico: funciones superiores conservadas, sin signos de foco motor o sensitivo.
1 Padre fallecido de cáncer de pulmón.
2 Esposo fallecido de tuberculosis pulmonar hace 15 años.
Hábitos:
1 Alcohol: niega
2 Tabaco: refiere 40 a 50 cigarrillos por día durante 30 años, con abandono hace 13 años
3 Otra medicación: cilostazol 100 mg/día, ranitidina 150 mg/12 hs, carbonato de calcio y diclofenac 50 mg/8 horas
Examen físico:
1 Vigil y orientada globalmente
2 Signos vitales: PA 130/70 mmHg; FC 75 lpm; FR 12 rpm; Temperatura 36,5 º C
3 Cabeza y cuello: conjuntivas pálidas, escleras blancas. No se palpan adenopatías ni tiroides.
4 Tórax: mamas sin tumoraciones ni lesiones evidentes.
5 Aparato respiratorio: expansión y excursión conservadas. Murmullo vesicular disminuido en forma generalizada con rales crepitantes en base izquierda.
6 Aparato cardiovascular: ruidos normofonéticos, soplo sistólico 3/6 en foco aórtico con irradiación al mesocardio y cuello. Pulsos periféricos conservados.
7 Abdomen: blando, depresible e indoloro. Hígado palpable 2 cm por debajo del reborde costal.
8 Miembros: desviación y deformidad a nivel de las falanges distales. Dolor a la movilización activa y pasiva de ambos hombros, codos, muñecas, dedos de las manos, rodillas y tobillos. No se palpan adenopatías ni edema.
9 Neurológico: funciones superiores conservadas, sin signos de foco motor o sensitivo.
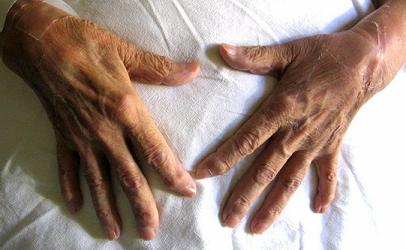
Fotografía 1: Desviación y deformidad a nivel de las falanges distales
Estudios complementarios:
Laboratorio general
|
1 mes previo
|
Ingreso
|
4to día
|
15to día
|
Hematocrito (%)
|
27
|
22
|
26
|
25
|
Hemoglobina (mg/dL)
|
8,7
|
6,9
|
8,2
|
8,1
|
Glóbulos blancos/mm3
|
1.600(47% Neutrófilos)
|
1.800
|
1.070
|
1.700
|
Plaquetas/mm3
|
463.000
|
.
|
.
|
295.000
|
Glicemia (mg/dL)
|
.
|
134
|
.
|
91
|
Urea (mg/dL)
|
70
|
89
|
94
|
44
|
Creatinina (mg/dL)
|
1,5
|
2,17
|
1,65
|
1,3
|
Natremia (mEq/L)
|
.
|
143
|
145
|
140
|
Potasemia (mEq/L)
|
.
|
3,9
|
4,7
|
3,9
|
Bilirrubina total (mg/dL)
|
.
|
.
|
.
|
.
|
ASAT (UI/L)
|
.
|
13
|
.
|
15
|
ALAT (UI/L)
|
.
|
15
|
.
|
9
|
FA (UI/L)
|
.
|
.
|
.
|
158
|
GGT (UI/L)
|
.
|
.
|
.
|
28
|
Colinesterasa (UI/L)
|
.
|
.
|
.
|
2.778
|
LDH (UI/L)
|
.
|
232
|
.
|
305
|
CPK (UI/L)
|
.
|
11
|
.
|
9
|
TP (segundos)
|
.
|
15.7
|
.
|
.
|
KPTT (segundos)
|
.
|
52
|
.
|
50
|
Albúmina (g/dl)
|
.
|
3
|
.
|
.
|
Proteínas (g/dL)
|
.
|
7,3
|
.
|
.
|
Uricemia (mg/dL)
|
.
|
10,9
|
.
|
.
|
Colesterol total (mg/dL)
|
117
|
75
|
.
|
.
|
Colesterol HDL (mg/dL)
|
28
|
.
|
.
|
.
|
Colesterol LDL (mg/dL)
|
64
|
.
|
.
|
.
|
Triglicéridos (mg/dL)
|
126
|
98
|
.
|
.
|
VES (mm/ 1ºhora)
|
84
|
.
|
.
|
.
|
Fotografía 1: Desviación y deformidad a nivel de las falanges distales.
1 Estado ácido base (12vo día): FiO2 21%. pH 7,37, pCO2 31 mmHg, pO2 92 mmHg, EB -6 mmol/L, HCO3 18 mmol/L, saturación Hb 97%.
2 Orina completa (12vo día, sin anticoagulación): hemoglobina ++++, campo cubierto de hematíes, leucocitos aislados.
3 Sedimento urinario (14to día): campo cubierto de hematíes eumórficos
1 Electrocardiograma (ingreso): Ritmo sinusal, frecuencia cardíaca 80 lpm, PR 0,12”, qRs 0,08”, QT 0,40”, aqRs +60º, infradesnivel del ST DI – DIII – V3 a V6 (necrosis inferior).
2 Radiografía de tórax (ingreso): relación cardio-torácica conservada, calcificación del anillo aórtico, fondos de saco pleurales libres, sin imágenes de condensación pulmonar
2 Orina completa (12vo día, sin anticoagulación): hemoglobina ++++, campo cubierto de hematíes, leucocitos aislados.
3 Sedimento urinario (14to día): campo cubierto de hematíes eumórficos
1 Electrocardiograma (ingreso): Ritmo sinusal, frecuencia cardíaca 80 lpm, PR 0,12”, qRs 0,08”, QT 0,40”, aqRs +60º, infradesnivel del ST DI – DIII – V3 a V6 (necrosis inferior).
2 Radiografía de tórax (ingreso): relación cardio-torácica conservada, calcificación del anillo aórtico, fondos de saco pleurales libres, sin imágenes de condensación pulmonar
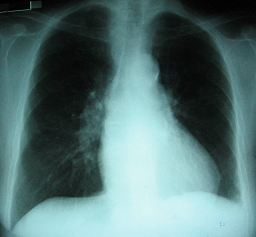
Radiografía de tórax de frente (ingreso): relación cardio-torácica conservada, calcificación del anillo aórtico, fondos de saco pleural libres, sin imágenes de condensación pulmonar .
Frotis de sangre periférica
|
2do día
|
5to día
|
6to día
|
Hematocrito (%)
|
27
|
26
|
27
|
Glóbulos blancos/mm3
|
800
|
1.600 (448 nt)
|
1.600 (900 nt)
|
Plaquetas/mm3
|
200.000
|
300.000
|
450.000
|
Microcitos, macrocitos, normocromia, macroplaquetas
3 Test de Coombs directo: positivo día 14, negativo día 15 (distintos operadores y reactivos)
4 Serologías
4 Serologías
- VDRL, VIH y VHC: no reactivas
5 Ecocardiograma bidimensional (2do día): Ventrículo izquierdo no dilatado, con hipertrofia leve, sin alteración de la motilidad. Función sistólica del ventrículo izquierdo normal. Raíz aórtica de características normales. Dilataciónleve de aurícula izquierda. Ecograma valvular normal, con cavidades derechas y pericárdicas sin particularidades.
6 Ecografía abdominal (6to día): Esplenomegalia (diámetro longitudinal 135 mm) de parénquima homogéneo. Riñón izquierdo con disminución del espesor cortical (6 mm), sin diferenciación córtico medular.
7 TC tórax alta resolución (9no día): engrosamiento de septos en forma bilateral y engrosamiento peribronquial bilateral a predominio central.
6 Ecografía abdominal (6to día): Esplenomegalia (diámetro longitudinal 135 mm) de parénquima homogéneo. Riñón izquierdo con disminución del espesor cortical (6 mm), sin diferenciación córtico medular.
7 TC tórax alta resolución (9no día): engrosamiento de septos en forma bilateral y engrosamiento peribronquial bilateral a predominio central.
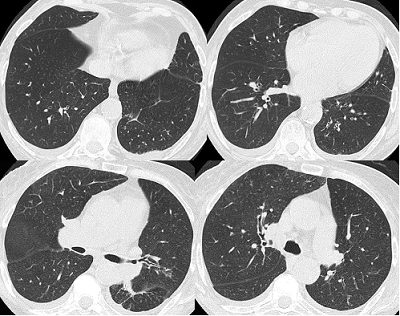
TC de tórax alta resolución (9no día): engrosamiento de septos en forma bilateral y engrosamiento peribronquial bilateral a predominio central
8 Extendido de médula ósea: médula hipercelular con progenie mieloide y eritroide bien representadas, con discretos cambios megaloblásticos. En la serie mieloide impresiona bloqueo madurativo a nivel de metamielocitos y de neutrófilos en cayado.
9 Estudios microbiológicos
9 Estudios microbiológicos
- Cultivo de esputo para gérmenes comunes (1er día): se observan colonias de Streptococcus viridans y de Pseudomona aeruginosa, siendo esta última sensible a amikacina, ceftazidima, cefepime, meropenem, ciprofloxacina, imipenem, piperacilina – tazobactam.
- Hemocultivos (2do día) negativos.
- Hemocultivos (14to día) negativos.
- Urocultivo (14to día) negativo.
- Esputo para bacilos ácido alcohol resistentes: 3 muestras negativas (1er, 2do y 3er días).
- Esputo para gérmenes comunes (9no día): se observan colonias de Klebsiella pneumoniae sensible a amikacina, gentamicina, imipenen, meropenem, cotrimoxazol, piperacilina – tazobactam y tigeciclina; resistente a ampicilina, cefalotina, ceftazidima y cefotaxime.
Evolución:
Al ingreso a sala general (4to día), por persistencia de fiebre y sospecha de neumonía se continúa con ampicilina y se amplía el espectro con ceftazidima – amikacina al interpretar a la paciente como neutropénica febril. Respecto a los estudios microbiológicos, no se encontraron hallazgos positivos.
Continúa la internación presentando 1 registro febril cada 24 – 48 hs. Al día 14to se constata una flebitis en el antebrazo izquierdo por lo que inicia tratamiento con vancomicina, previa toma de cultivos. Al día siguiente, por persistir febril se suspende ceftazidima – amikacina para reevaluación infectológica.
Respecto a los parámetros de laboratorio, se evidencia con el transcurso de la internación una mejoría en la función renal, con persistencia de la bicitopenia.
Al ingreso a sala general (4to día), por persistencia de fiebre y sospecha de neumonía se continúa con ampicilina y se amplía el espectro con ceftazidima – amikacina al interpretar a la paciente como neutropénica febril. Respecto a los estudios microbiológicos, no se encontraron hallazgos positivos.
Continúa la internación presentando 1 registro febril cada 24 – 48 hs. Al día 14to se constata una flebitis en el antebrazo izquierdo por lo que inicia tratamiento con vancomicina, previa toma de cultivos. Al día siguiente, por persistir febril se suspende ceftazidima – amikacina para reevaluación infectológica.
Respecto a los parámetros de laboratorio, se evidencia con el transcurso de la internación una mejoría en la función renal, con persistencia de la bicitopenia.
El 15to día presentó un segundo evento coronario agudo, con cambios en el ECG sin elevación de enzimas cardíacas, por lo que ingresa nuevamente a Unidad Coronaria. A las 48 horas vuelve a la sala general, luego de haber recibido tratamiento anticoagulante EV y con nitratos, con buen estado general.
Estudios recibidos durante la internación:
Estudios recibidos durante la internación:
1 Ac anti leucocitarios: negativos.
2 Ac anti cardiolipinas: IgM negativa; IgG positiva a títulos altos
3 Orina de 24 hs:
2 Ac anti cardiolipinas: IgM negativa; IgG positiva a títulos altos
3 Orina de 24 hs:
- Proteínas 1,06 g/24h
- Clearence de creatinina 21 ml/min
4 Serología para Parvovirus B19: IgM negativa
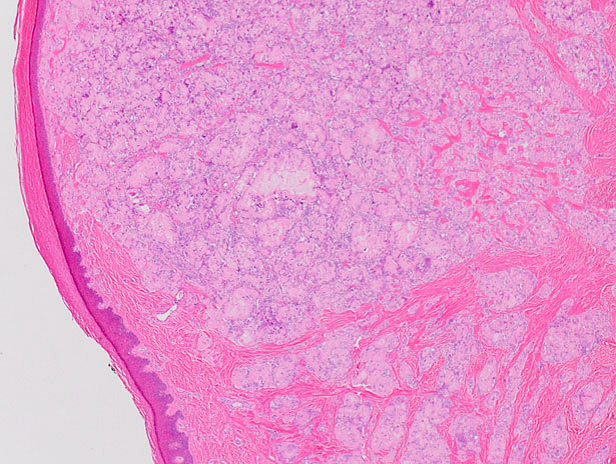
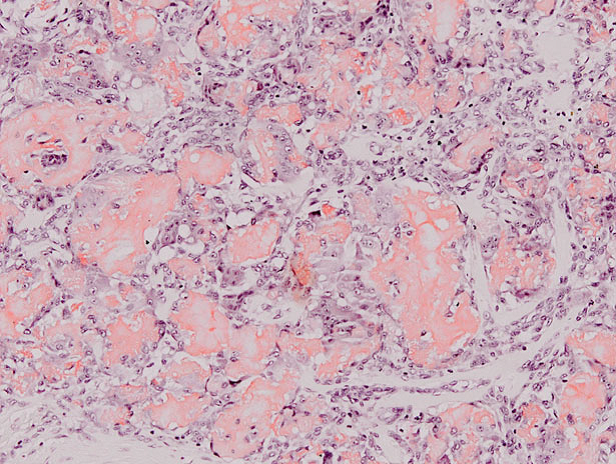
5 Anatomía patológica de médula ósea: médula levemente hipercelular con buena representación de las 3 progenies
6 Citología de orina: negativa para células neoplásicas
5 Anatomía patológica de médula ósea: médula levemente hipercelular con buena representación de las 3 progenies
6 Citología de orina: negativa para células neoplásicas
Resultados de estudios pendientes:
Discusión del caso clínico
Evolución del caso clínico
Laboratório inmunológico:
o Factor reumatoideo (látex): reactivo 1/40
o Anti ADN: negativo
o FAN: positivo patrón moteado homogéneo anular 1/2560
o Proteinograma por electroforesis: leve aumento policlonal de inmunoglobulinas y discreta disminución de la albúmina
o Complemento: CH50: 17 UCH50 (25 a 48), C3: 89 mg/dL (102 a 145), C4: 19 mg/dL (20 a 50). Hipocomplementemia
o Ac anti Sm: negativo
o Anti ADN: negativo
o FAN: positivo patrón moteado homogéneo anular 1/2560
o Proteinograma por electroforesis: leve aumento policlonal de inmunoglobulinas y discreta disminución de la albúmina
o Complemento: CH50: 17 UCH50 (25 a 48), C3: 89 mg/dL (102 a 145), C4: 19 mg/dL (20 a 50). Hipocomplementemia
o Ac anti Sm: negativo
Discusión del caso clínico
Dra. María Victoria Ferretti - Residente de 3er año de Clínica Médica – Hospital Provincial del Centenario – Rosario
Se va a discutir el caso de una paciente mujer, de 76 años, que como antecedente es hipertensa, cardiópata, dislipémica, ex tabaquista, y con historia de artralgias de 20 a 30 años de evolución para lo cual ingería diclofenac en forma reglada. Se interna en nuestro hospital por un síndrome coronario agudo, constatándose a su vez un síndrome febril y en el laboratorio valores de bicitopenia e insuficiencia renal, agregando hematuria y dentro del laboratorio inmunológico un FAN positivo a título alto.
Se tomará como dato guía para la discusión la bicitopenia. Sabemos que las causas son múltiples y no van a ser desarrolladas todas, debido a que muchas pueden ser descartadas. Por ejemplo, la paciente no refiere haber estado expuesta a tóxicos o a fármacos capaces de inducir bicitopenia (sulfas, quimioterápicos, etc). Considerando las causas infecciosas, a pesar de presentar un foco positivo para tuberculosis, presenta tres esputos para BAAR negativos; y los métodos por imágenes disponibles no evidencian afección de ningún órgano de la economía por una enfermedad granulomatosa, lo que alejaría también una histoplasmosis. Por lo tanto, considero que las causas más importantes a desarrollar en nuestra paciente son las inmunológicas y las neoplásicas hematológicas. No así las neoplasias sólidas, debido a que no tenemos evidencia al examen físico ni por los métodos por imágenes de tumores; y además, los síndromes paraneoplásicos que producen los tumores sólidos cursan en su mayoría con proliferación de las células hematológicas, y menos frecuentemente con disminución de las mismas.
De esta manera, para comenzar a desarrollar las ENFERMEDADES INMUNOLÓGICAS que podrían justificar las alteraciones de nuestra paciente, es importante remarcar que el FAN presenta alta sensibilidad (cerca del 100%) para enfermedades inmunológicas sistémicas pero baja especificidad debido a que son múltiples las patologías que pueden elevarlo, tanto sistémicas como órgano-específicas. Dentro de las enfermedades inmunológicas que cursan con FAN elevado, desarrollaré el lupus eritematoso sistémico (LES), artritis reumatoidea (AR) y la enfermedad mixta del tejido conectivo (EMTC).
El Lupus Eritematoso Sistémico, como sabemos, en una enfermedad sistémica mediada por autoanticuerpos e inmunocomplejos dirigidos contra las propias células del organismo. Para su diagnóstico deben presentarse 4 ó más criterios, de los 11 estipulados:
1 Eritema malar
2 Eritema discoide
3 Fotosensibilidad
4 Úlceras orales
5 Serositis (pleuritis o pericarditis)
6 Artritis (no erosiva, de 2 ó más articulaciones periféricas)
7 Trastornos neurológicos (convulsiones o psicosis)
8 Trastornos renales (proteinuria o cilindros celulares)
9 Trastornos hematológicos (anemia, leucopenia, trombocitopenia)
10 Trastornos inmunes (Anti ADN, Anti Sm, anticuerpos antifosfolípidos)
11 FAN positivo
2 Eritema discoide
3 Fotosensibilidad
4 Úlceras orales
5 Serositis (pleuritis o pericarditis)
6 Artritis (no erosiva, de 2 ó más articulaciones periféricas)
7 Trastornos neurológicos (convulsiones o psicosis)
8 Trastornos renales (proteinuria o cilindros celulares)
9 Trastornos hematológicos (anemia, leucopenia, trombocitopenia)
10 Trastornos inmunes (Anti ADN, Anti Sm, anticuerpos antifosfolípidos)
11 FAN positivo
He remarcado los que presentaría nuestra paciente. En cuanto a las artritis, en el LES son varias las formas de afección articular, siendo la más frecuente la presentación con artralgias y artritis en forma intermitente. Nuestra paciente refiere rigidez matinal, que podría ser explicada por tendinitis de la mano, que se manifestaría clínicamente como una pseudo – rigidez.
Como manifestación renal, tanto la proteinuria como la hematuria con eritrocitos dismórficos podrían ser explicadas por una nefropatía lúpica.
Tenemos los anticuerpos anticardiolipinas positivos a título alto, como así también el FAN.
Tenemos los anticuerpos anticardiolipinas positivos a título alto, como así también el FAN.
Considerando la bicitopenia, en el LES la misma se explicaría por varios mecanismos, dependiendo de la serie celular afectada.
La anemia suele ser normocítica normocrómica y puede ser debida a la propia enfermedad crónica; a pérdidas digestivas, debido generalmente a los AINES o corticoides utilizados por estos pacientes; a insuficiencia renal; a hemólisis, que no parece ser el caso de nuestra paciente, debido a que los signos indirectos de hemólisis de laboratorio son negativos, como así también el Test de Coombs; puede ser secundaria a infecciones debido a inmunodepresión o secundaria a drogas; y con menos frecuencia puede deberse a hiperesplenismo, mielofibrosis, mielodisplasia o aplasia de la médula ósea.
La leucopenia (<4500/mm3), manifestada más frecuentemente en forma de linfopenia (<1500/mm3), suele deberse a mecanismos mediados por linfocitos T supresores o por anticuerpos anti linfocitos, que en nuestra paciente son negativos. La neutropenia suele mostrar valores menores a 1000/mm3 y es menos frecuente. Suele deberse a mecanismos inmunológicos o a drogas. En ocasiones, debido al uso crónico de corticoides, puede verse disminución de eosinófilos y de basófilos.
También dentro de las alteraciones hematológicas se describen las adenopatías, la esplenomegalia y la VES aumentada, siendo las dos últimas alteraciones reflejo de actividad de la enfermedad.
Es importante remarcar que está descripto un aumento de la incidencia deinfarto agudo de miocardio, isquemia cerebral o apoplejía en pacientes lúpicas, fundamentalmente en aquellas que presentan positivos los anticuerpos antifosfolipidos. Esta complicación se presentaría luego de 15 a 20 años de evolución de la enfermedad, y la cardiopatía isquémica en estos pacientes se explicaría por un mecanismo de aterosclerosis acelerada. De esta manera, se ha postulado que a los factores de riesgo clásicos de cardiopatía isquémica, como ser la hipertensión arterial, la dislipemia, la edad avanzada, la diabetes, se sumarían los factores de riesgo relacionados a la enfermedad lúpica, como ser la inflamación de la propia enfermedad, la presencia de autoanticuerpos dirigidos contra el endotelio, HDL y antifosfolipidos, los inmunocomplejos circulantes y la presencia de nefritis e incluso dislipemia producida por la propia enfermedad; todos ellos en suma actuarían como factores aceleradores de aterosclerosis en estos pacientes.
Este fenómeno ha llevado a la formulación de múltiples y variados estudios, uno de ellos del año 2007, postula que tanto la edad avanzada al diagnóstico de la enfermedad, la larga evolución de la misma y los niveles altos de homocisteína actuarían como factores independientes relacionados con la progresión de la aterosclerosis, sugiriendo por lo tanto un control agresivo del LES en estos pacientes para retrasar el progreso de la aterosclerosis.
Este fenómeno ha llevado a la formulación de múltiples y variados estudios, uno de ellos del año 2007, postula que tanto la edad avanzada al diagnóstico de la enfermedad, la larga evolución de la misma y los niveles altos de homocisteína actuarían como factores independientes relacionados con la progresión de la aterosclerosis, sugiriendo por lo tanto un control agresivo del LES en estos pacientes para retrasar el progreso de la aterosclerosis.
A su vez, recordemos que nuestra paciente tiene 76 años, y lo más frecuente es que el lupus se presente entre la segunda y tercera década de la vida. Sin embargo, se ha visto en la edad avanzada, y alcanza una incidencia del 12 – 18%. Se caracteriza por presentar un curso más benigno y debido a que suele presentarse con manifestaciones inespecíficas y en forma solapada, el diagnóstico se realiza en forma tardía.
En base a los estudios realizados en pacientes de edad avanzada, se ha visto que las manifestaciones clínicas no son iguales que en edades tempranas. Se ha descripto mayor incidencia de artromialgias, serositis, compromiso pulmonar, positividad para FR, Anti Ro, Anti La y FAN. Con menor frecuencia se han reportado manifestaciones cutáneas, artritis, nefritis, hipocomplementemia y Anti RNP.
En cuanto a la nefritis, manifestada como aumento de la creatininemia y sedimento patológico (proteinuria y hematuria con eritrocitos dismórficos), suele verse agravada o precipitada por enfermedades sistémicas comunes a esta edad, como la hipertensión arterial y la diabetes mellitus. Cabe remarcar que la progresión a la insuficiencia renal crónica se observa en un 8 – 18% y la mortalidad debida a nefropatía alcanza un 25%.
A favor de lupus eritematoso sistémico (LES) tenemos la bicitopenia, los eventos isquémicos, la VES acelerada, la esplenomegalia, la proteinuria, la hematuria, la hipocomplementemia, el FAN positivo y la positividad de los anticuerpos antifosfolipidos. En contra tenemos la edad pero, como mencionamos, puede verse en la edad avanzada, por lo tanto considero que el LES es un diagnóstico probable en nuestra paciente.
En cuanto a la artritis reumatoidea (AR) sabemos que es una enfermedad sistémica, de etiología desconocida y que afecta preferentemente a mujeres (3:1). Se requieren 4 de los 7 criterios diagnósticos para establecer el mismo. Nuestra paciente presentaría la artritis simétrica de las articulaciones de las manos, codos, muñecas, hombros, rodillas y tobillos, con rigidez matinal, y la positividad de factor reumatoideo. Pero recordemos que la artritis debe ser erosiva, y no se han encontrado erosiones en las radiografías de manos de nuestra paciente. A nivel hematológico, la AR suele cursar con anemia normocítica, normocrómica, y es rara la leucopenia pero puede observarse, y ser consecuencia del uso continuo de drogas como AINES o corticoides.
Una de las complicaciones de la AR que debo considerar es el Síndrome de Felty, que suele presentarse en pacientes con FR positivo y que se caracteriza por la presencia de esplenomegalia y granulocitopenia. Esta granulocitopenia se debería a un disbalance entre la producción de los granulocitos en la médula ósea y su remoción a nivel de la circulación periférica, y que se vería aumentada debido fundamentalmente a mecanismos inmunológicos (inmunocomplejos o anticuerpos anti granulocitos) o a anormalidades de la médula ósea. Lo característico es encontrar en la histología una médula ósea hipercelular; puede observarse bloqueo madurativo, como presentaba nuestra paciente.
Y es preciso diferenciar bien a los síndromes indiferenciados del tejido conectivo, de los síndromes de superposición. De esta forma, los síndromes indiferenciados son aquellos en los cuales un paciente presenta síntomas, signos o datos de laboratorio sugestivos de una enfermedad del tejido conectivo, pero sin llegar a cumplir los criterios diagnósticos de la misma. En cambio, los síndromes de superposición son aquellos en los cuales el paciente presenta dos o más enfermedades del tejido conectivo definidas. Dentro de este último grupo, una de las más estudiadas ha sido la superposición entre LES y AR, también conocida como “Rhupus”, que fue descripta en 1971 debido a que era frecuente observar que mujeres con historia de 15 – 20 años de AR comenzaron a presentar características clínicas de LES. Así se observó también que las pacientes con AR pueden presentar en el 40% de los casos FAN positivo y, a la inversa, 40% de las pacientes con LES pueden cursar con FR positivo. Considero que nuestra paciente presenta muchas características clínicas de LES, y que para AR presentaría la rigidez matinal, la afectación de las manos, la artritis simétrica y la positividad del FR. La superposición de ambas enfermedades constituiría un diagnóstico tentador, pero alejado en nuestra paciente, debido a que todo podría explicarse por una sola enfermedad como que sería el LES.
En cuanto a la enfermedad mixta del tejido conectivo (EMTC), es un síndrome de superposición entre LES, AR, polimiositis y esclerodermia, que afecta a mujeres jóvenes dando artritis no erosiva de las manos, con deformidades, edema y Raynaud. Se caracteriza por la positividad del anticuerpo anti RNP U1. Las alteraciones hematológicas comprenden anemia, generalmente hemolítica, y es rara la leucopenia. El FR es positivo en el 50% de los casos y se observa también hipergammaglobulinemia. Considero que, debido a que la paciente no presenta características de polimiositis ni de esclerodermia, ni otras alteraciones características de esta entidad, la EMTC correspondería a un diagnóstico alejado.
A su vez no debemos olvidar que la paciente es hipertensa, y que la nefropatía hipertensiva, mediante nefroesclerosis, puede causar isquemia de túbulos y glomérulos, dando elevación de la creatininemia, hematuria microscópica y proteinuria leve.
También quiero considerar brevemente el consumo crónico de AINES. El diclofenac (aunque raramente), puede causar disminución de todas las series hematopoyéticas, considerando que la anemia suele ser hemolítica o aplásica. En la última actualización de la American Heart Association del año 2007 se ha descripto un aumento del riesgo cardiovascular, principalmente de infarto agudo de miocardio (IAM), en personas que consumen los mismos en forma diaria y crónica.
Recordemos también que los AINES pueden causar nefropatía, en forma de nefritis intersticial o de necrosis papilar, manifestándose clínicamente como insuficiencia renal y hematuria micro o macroscópica debido a la eliminación de las papilas necróticas. Esto se presentaría en pacientes que consumen 2 o más AINES en forma diaria y por más de 5 años. Recordemos que nuestra paciente consumía diclofenac y aspirina. Considero que tal vez la suma de la toxicidad producida por la aspirina y el diclofenac podría haber actuado como factor agravante de las alteraciones hematológicas, renales y cardiovasculares de nuestra paciente. Cabe aclarar que debido a que estos pacientes suelen presentar riesgo cardiovascular por otros factores, no estaría contraindicado el uso de aspirina a bajas dosis.
En cuanto a las NEOPLASIAS HEMATOLÓGICAS, tanto las leucemias, los síndromes linfoproliferativos y los mielodisplásicos podrían explicar la pérdida de peso referida por la paciente, como así también la bicitopenia. Pero la paciente presenta otras alteraciones, como ser las manifestaciones renales y articulares, que difícilmente podrían ser explicadas por estas entidades. A su vez, la paciente presenta claramente un contexto autoinmune. Y tenemos un extendido de médula ósea que no muestra células neoplásicas ni evidencia de mielodisplasia, con un informe verbal de anatomía patológica de la médula ósea que es negativa para células neoplásicas. Por lo tanto, estas entidades estarían prácticamente descartadas en nuestra paciente.
Como conclusión, considero que estamos ante la presencia de una enfermedad eminentemente inmunológica con compromiso hematológico, renal y articular; pudiendo corresponder preferentemente a un Lupus Eritematoso Sistémico. Dejaría en un segundo plano un síndrome de superposición, como el Rhupus. Y considero también, que la nefropatía podría haberse visto agravada por el consumo crónico de AINES y por la hipertensión arterial.
Sugeriría como conducta completar el laboratorio inmunológico, fundamentalmente los anticuerpos nucleares extraíbles (ENA). Considero importante la realización de una biopsia renal, con el fin de estadificar la nefropatía, si pensamos en un LES, y de esta forma considerar la implementación o no de terapia inmunosupresora. Por lo tanto, aguardaría el resultado de la biopsia para iniciar corticoides. Luego de la biopsia iniciaría anticoagulación por la posibilidad de un síndrome antifosfolípidos. A su vez, debido a la severidad de las artralgias, considero importante la implementación de una droga modificadora de la enfermedad, como ser la hidroxicloroquina. Suspendería el diclofenac y otros AINES, excepto la aspirina. Consideraría prudente la realización de un fondo de ojo, para evaluar daño de órgano blanco. Y planteo el interrogante en cuanto a la posibilidad de realización de una cinecoronariografía, como estudio de su cardiopatía isquémica, que constituiría el componente más severo actualmente en nuestra paciente.
Sugeriría como conducta completar el laboratorio inmunológico, fundamentalmente los anticuerpos nucleares extraíbles (ENA). Considero importante la realización de una biopsia renal, con el fin de estadificar la nefropatía, si pensamos en un LES, y de esta forma considerar la implementación o no de terapia inmunosupresora. Por lo tanto, aguardaría el resultado de la biopsia para iniciar corticoides. Luego de la biopsia iniciaría anticoagulación por la posibilidad de un síndrome antifosfolípidos. A su vez, debido a la severidad de las artralgias, considero importante la implementación de una droga modificadora de la enfermedad, como ser la hidroxicloroquina. Suspendería el diclofenac y otros AINES, excepto la aspirina. Consideraría prudente la realización de un fondo de ojo, para evaluar daño de órgano blanco. Y planteo el interrogante en cuanto a la posibilidad de realización de una cinecoronariografía, como estudio de su cardiopatía isquémica, que constituiría el componente más severo actualmente en nuestra paciente.
Evolución del caso clínico
Dr. Eduardo Gonzalez – Residente de 2do año de Clínica Médica – Hospital Provincial del Centenario – Rosario
Al 17mo día de internación inicia tratamiento con factor estimulante de colonias de granulocitos, con incremento del recuento de glóbulos blancos. Por otro lado, se evidencia nuevo deterioro de la función renal con cifras de creatinininemia cercanas a los 2,3 mg/dL, que mejora levemente con el aporte de cristaloides en el transcurso de los días.
Al 25to día de internación se encuentra neutropénica (glóbulos blancos 500/mm3), con insuficiencia renal (creatininemia 1,9 mg/dL), buen estado general, sin repetición de eventos coronarios.
Se realiza punción biopsia renal, procedimiento bien tolerado y sin complicaciones. Continúa con leucopenia, y debido a la aparición en pulpejos de dedos de ambas manos de lesiones compatibles con vasculitis, se decide realizar pulso de corticoides (metilprednisolona 1000 mg día durante 3 días consecutivos). El mismo es bien tolerado, sin producirse durante su realización complicaciones de tipo cardiovascular ni del medio interno.
Posterior a este tratamiento se evidencia una mejoría significativa de las artralgias y del estado general, que se acompañan de aumento del recuento de los glóbulos blancos y de mejoría en los parámetros de laboratorio renal.
El resultado de la biopsia renal informa glomerulonefritis lúpica mesangial, dato que confirma el diagnóstico de lupus eritematoso sistémico.
Respecto a la función renal, se evidencia incremento en la falla que mejora con el aporte de líquidos sin llegar a valores normales. Se otorga el alta hospitalaria con indicaciones de continuar tratamiento con corticoides (prednisona 0,5 mg/kg/día) con control por consultorio externo de Clínica Médica, Reumatología y Nefrología.
Diagnóstico final: Lupus Eritematoso Sistémico
Diagnóstico final: Lupus Eritematoso Sistémico
BIBLIOGRAFIA
· Bevra H. “Lupus eritematoso generalizado”. Principios de Medicina Interna; 2005: 2158 – 2166.
· Peter E. “Artritis reumatoidea”. Principios de Medicina Interna; 2005: 2166 – 2175.
· Banwari S. “Rhupus: report of 3 cases”. J Indian Rheumatol Assoc 2003; 11: 51 – 54
· Bennet R. “Clinical manifestations of mixed connective tissue disease”. In Up To Date, Nov 2006.
· Schur P. “Diagnosis and differential diagnosis of systemic lupus erythemotosus in adults”. In Up To Date, Ene 2007.
· Kathleen M et al. “Predictors of Carotid Atherosclerosis in Systemic Lupus Erythematosus”. The Journal of Rheumatology, 2006; 33:2458-63
· Naomi F. et al. “Systemic Lupus Erythematosus Evolving Into Rheumatoid Arthritis”. Te Journal of Rheumatology, 2006;33:188-90.
· Bevra H. “Systemic Lupus Erythematosus and Accelerated Atherosclerosis”. The New England Journal of Medicine 2003; 2379 – 2380.
· Marc E et al. “Analgesic Nephropathy”. The New England Journal of Medicine; 1998: 446 – 452.
· Alarcón G. “Síndromes indiferenciados y de superposición del tejido conectivo”. Reviste Mexicana de Reumatología, 2002; 17 (3): 199 - 205
· Elliot, M. “Use of Nonsteroidal Antiinflammatory Drugs. An Update for Clinicians”. Circulation; Mar 2007; 115: 1634 – 1642
· Cohen M, et al. “Concurrence of Rheumatoid Arthritis and Systemic Lupus Erythematosus: report of 11 cases”. Annals of the Rheumatic Diseases, 1987; 46, 853 – 858
· Roman MJ et al. “Rate and determinants of progression of atherosclerosis in systemic lupus erythematosus”. Arthritis Rheum. 2007 Oct;56(10):3412-9
· Rovensky J. “Systemic Lupus Erythematosus in the Elderly”. Autoinmunity Reviews, 235-239. Dec 2008
· Bevra H. “Lupus eritematoso generalizado”. Principios de Medicina Interna; 2005: 2158 – 2166.
· Peter E. “Artritis reumatoidea”. Principios de Medicina Interna; 2005: 2166 – 2175.
· Banwari S. “Rhupus: report of 3 cases”. J Indian Rheumatol Assoc 2003; 11: 51 – 54
· Bennet R. “Clinical manifestations of mixed connective tissue disease”. In Up To Date, Nov 2006.
· Schur P. “Diagnosis and differential diagnosis of systemic lupus erythemotosus in adults”. In Up To Date, Ene 2007.
· Kathleen M et al. “Predictors of Carotid Atherosclerosis in Systemic Lupus Erythematosus”. The Journal of Rheumatology, 2006; 33:2458-63
· Naomi F. et al. “Systemic Lupus Erythematosus Evolving Into Rheumatoid Arthritis”. Te Journal of Rheumatology, 2006;33:188-90.
· Bevra H. “Systemic Lupus Erythematosus and Accelerated Atherosclerosis”. The New England Journal of Medicine 2003; 2379 – 2380.
· Marc E et al. “Analgesic Nephropathy”. The New England Journal of Medicine; 1998: 446 – 452.
· Alarcón G. “Síndromes indiferenciados y de superposición del tejido conectivo”. Reviste Mexicana de Reumatología, 2002; 17 (3): 199 - 205
· Elliot, M. “Use of Nonsteroidal Antiinflammatory Drugs. An Update for Clinicians”. Circulation; Mar 2007; 115: 1634 – 1642
· Cohen M, et al. “Concurrence of Rheumatoid Arthritis and Systemic Lupus Erythematosus: report of 11 cases”. Annals of the Rheumatic Diseases, 1987; 46, 853 – 858
· Roman MJ et al. “Rate and determinants of progression of atherosclerosis in systemic lupus erythematosus”. Arthritis Rheum. 2007 Oct;56(10):3412-9
· Rovensky J. “Systemic Lupus Erythematosus in the Elderly”. Autoinmunity Reviews, 235-239. Dec 2008






.png)




{kind=link}